
Abstract
Background
Jaundice in the newborn period is a very common entity; rare inherited causes are often forgotten. Persistent unconjugated hyperbilirubinemia in the intermediate levels with non-hemolytic features must prompt the necessity for evaluating for genetic defects in bilirubin metabolism.
Case presentation
Three-and-a-half-month-old first-born girl of consanguineous marriage presented with jaundice from day 5 of life. Dark yellow color urine or pale stools were not present. Antenatal and birth history was normal. She had mild pallor and icterus and no hepatosplenomegaly. Total serum bilirubin was 8.2 mg/dl, and direct was 0.4 mg/dl. Workup for hemolytic anemia, thyroid function test, and sonography of abdomen was normal. Syrup phenobarbitone was started, and bilirubin levels after dropped to 2 mg/dl. Crigler-Najjar type II syndrome (CN II) or Gilbert’s syndrome (GS) was suspected. Next-generation sequencing for UGT1A1 gene mutation showed homozygous missense mutation consistent with CN II and 7 TA repeats in the promoter region consistent with GS. Bilirubin levels gradually fell after starting oral phenobarbitone syrup, and at 5 years of age, a trial of withholding phenobarbitone was given, and bilirubin levels remained lower, and she is asked to follow-up with bilirubin levels every 15 days to assess the need for reintroducing the therapy. Parents are planning for a second pregnancy, and a preconception genetic counseling has been done.
Conclusion
Genetic confirmation of coexistence of mutations causing GS and CN II have an implication on long-term neurological complications of unconjugated hyperbilirubinemia in stress or crisis situations. Prenatal diagnostic testing must be advised for detecting homozygous UGT1A1 mutations to diagnose CN II and Gilbert mutations for each of the future pregnancies. Considering the side effects of long-term phenobarbitone therapy, the decision can be taken on case-to-case basis of stopping the therapy while monitoring TSB levels.
Similar content being viewed by others


Background
Pathological hyperbilirubinemia in neonatal period can be classified into unconjugated and conjugated hyperbilirubinemias [1]. The indirect hyperbilirubinemias mainly comprise groups of hemolytic anemias or disorders of bilirubin metabolism. JF Crigler and VA Najjar in 1952 studied seven patients with congenital familial non-hemolytic jaundice and kernicterus and proved that the abnormality was inherited most likely in an autosomal recessive manner [2]. Crigler-Najjar syndrome is a rare genetic disease with an estimated incidence of 0.6–1.0 per million live births around the world [3], and Gilbert’s syndrome (GS) is one of the commoner genetic but benign cause of unconjugated hyperbilirubinemia worldwide with a population frequency of 5–10% [4]. Uridine diphospho-glucuronosyltransferase (UDPGT) is the primary enzyme needed for bilirubin glucuronidation in the liver and the gene UGT1A1 coding for it is located on the long (q) arm of chromosome 2 at position 37.1 [5], mutations of which causes abnormal accumulation of the unconjugated bilirubin which can cause bilirubin induced neurotoxicity (BIND) [1]. The complete absence of the enzyme activity (stop codon or frameshift mutation) causes Crigler-Najjar I syndrome (OMIM: 218,800) and decreased activity causes CN II (homozygous missense mutation) (OMIM: 606,785) [6]. CN II usually presents in infants or children with intermediate hyperbilirubinemia with bilirubin levels ranging from 1.5 to 22 mg/dl [6]. They usually respond well to oral lifelong phenobarbitone therapy. Gilbert’s syndrome (GS) (OMIM: 143,500) is a quite common benign condition which shows up in adolescence caused by polymorphisms in the promoter region of UGT1A1 gene (7 tandem repeats) which reduces its expression and decreases the enzyme activity. The bilirubin levels reach up to 3 mg/dl and usually does not require any treatment [7]. We present a case of coexistence of both Crigler-Najjar syndrome and Gilbert’s syndrome mutations in an infant presenting with indirect hyperbilirubinemia.
Case presentation
Three-and-a-half-month-old full-term baby girl born to third-degree consanguineous parents presented with yellow discoloration of the eyes and skin since day 5 of life. The parents hail from the western central part of India. Antenatal and birth history was uneventful. There was no history of clay-colored stools or dark yellow urine, bleeding manifestations, lethargy, seizures, and blood or blood product transfusion. Her family history was not significant. She was exclusively breastfed. She had developed a social smile, could recognize her mother, could hold her neck well and make cooing sounds, used to quieten when spoken to, and could visualize toys and reach out for them. On general examination, the baby was active and alert, weight at presentation was 5.5 kg, height was 62 cm, head circumference was 39 cm, and as per WHO growth chart for girls (0 to 5 years), she was on 0 SD. She had icterus and mild pallor. There was no obvious dysmorphisms or any other signs of liver cell failure. The liver was 2 cm palpable (span normal) with no free fluid. Thus, we had a 3.5-month-old well grown, developmentally normal, girl child with jaundice since day 5 of life, with no organomegaly, no facial dysmorphisms, and normal family history. She received double surface phototherapy in newborn period for 4 days after which the jaundice reduced on day 10 just to appear again on day 15. The child was readmitted for phototherapy once again at day 20. The jaundice reduced clinically on day 25 and reappeared on day 30 after which it was fairly persistent until 3 months of age when she presented to us. A complete blood count, bilirubin, and peripheral smear were done during this period which was within normal limits.
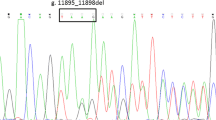
On presentation to us, the child had a total bilirubin of 8 mg/dl with indirect component of 7.6 mg/dl. The mother’s blood group was B positive, and the baby was O positive with direct and indirect Coombs test negative. Other liver function tests, complete blood count, urine routine examination, thyroid function tests, peripheral smear, reticulocyte count, sickling test, hemoglobin electrophoresis of parents, RBC membrane and enzyme studies, glucose six phosphate dehydrogenase enzyme levels, and ultrasonography of abdomen were all normal. Hence, inherited disorders of bilirubin metabolism were suspected, and a next-generation sequencing (NGS) of the child’s DNA sample was sent. Meanwhile, the child was started on oral phenobarbitone (5 mg/kg/day) after which there was a gradual decrease in jaundice (total bilirubin/indirect 4/3.6 mg/dl). The absence of kernicterus and hyperbilirubinemia responding to phenobarbitone favored a clinical diagnosis of CN II. NGS was done which revealed a homozygous 2 bp insertion of TA nucleotides [c.-55_-54insTA], leading to 7 TA repeats [A(TA)7TAA] in the promoter region of UGT1A1 gene known as UGT1A1*28 variation, which was pathogenic and another homozygous missense variation (chr2:234669097A > G; c.164A > G) in exon 1 of the UGT1A1 gene, that results in the amino acid substitution of arginine for histidine at codon 55 (p.His55Arg) which was pathogenic as was detected in this subject (Fig. 1). The variant analysis in Sanger sequencing was based on the UGT1A1 reference sequence ENST00000305208.5. This was consistent with the diagnosis of CN syndrome II and Gilbert’s syndrome (Table 1). The gene studies of the parents could not be done due to financial reasons. She was on daily oral phenobarbitone therapy (compliant) till the age of 5.5 years with bilirubin levels in the range of total bilirubin/indirect 2.6/1.49 mg/dl, and she is developmentally normal and growing well. The serial bilirubin values of the child are depicted in Table 2.

Sequence chromatogram and alignment to the reference sequence showing the variation in exon 1 of the UGT1A1 gene (chr2:234669097A > G;c.164A > G;p.His55arg) detected in homozygous condition in the baby
However, keeping in mind the effect of long-term phenobarbitone on the cognitive function and behavior of the child, a trial of withholding phenobarbitone therapy follows (total/indirect 3.6/2.4 mg/dl). She has been advised to repeat serum bilirubin levels every 2 weeks for the first few months and to restart phenobarbitone syrup if the TSB levels exceed 8 mg/dl. The parents are planning for a second pregnancy, and a preconception genetic counseling has been done.
Discussion
While clinically diagnosing CN II in an infant with bilirubin levels up to 20 mg/dl in the absence of Kernicterus is easier, diagnosing GS at this age without genetic studies is difficult. Comparison of genetic patterns of our patient along with other similar studies is elicited in Table 3.
Usually, children having Gilbert’s mutations remain asymptomatic, and the jaundice becomes apparent during early to late adolescence along with an intercurrent infection, stress, or crisis situation [11]. However, a combination with a Crigler-Najjar type 2 mutation could be the reason for early presentation of our child from neonatal period. Also, levels of total serum bilirubin (TSB) were thought to be around 3 mg/dl only in olden days; however, Gilbert’s syndrome in the current era presents with higher bilirubin levels which may confuse the picture. A large study (170 children with genetically proven Gilbert mutations) among the Indian population by Sood V et al. had 133 cases with homozygous and 37 with heterozygous status. The median serum total bilirubin (TB) levels were around 3.3 mg/dl with maximum levels reaching up to 18 mg/dl. The mean age of diagnosis in this study was around 13.6 years. They also showed that bilirubin levels were higher in heterozygous status than homozygous status [12].
The presence of Gilbert mutations along with homozygous mutations of UGT1A1 gene for CN II causes varied presentation in infants. Laura CozzI et al. reported two Chinese neonates who presented with prolonged severe jaundice > 15 mg/dl and found homozygous for a variant site within the coding region of the gene in the 4 exon, c.1091C > T, p. Pro364Leu. the P364L mutation. Jaundice was persistent and unresponsive to phototherapy. The infants however responded well to phenobarbitone, TSB levels gradually fell, and jaundice resolved in a few months, and the therapy was stopped. These infants had intermediate levels of TSB between Gilbert’s syndrome and Crigler-Najjar syndrome and responded completely to phenobarbitone therapy [13].
Studies have also shown that cases with both Gilbert and CN II mutations suffer from liver parenchymal changes, hence the need to identify this double mutation earlier. In a study from China, percutaneous liver biopsy of 59 patients of unconjugated hyperbilirubinemia was done for assessing inflammation and extent of fibrosis. They showed that the linked polymorphic mutations, A(TA)7TAA and c.-3279 T > G in UGT1A1, were most strongly associated with GS, whereas mutations in the coding region, especially p.G71R and p.Y486D, were more strongly associated with CNS-II. Iron deposition was more common in liver biopsies from patients with CNS-II than in those with GS [14].
After ruling out hemolytic conditions, the absence of kernicterus and response to phenobarbitone makes the clinical diagnosis of CN II in newborn period obvious; however, these children may still carry a GS mutation. Gailite L et al. described a 17-year-old boy born to non-consanguineous parents who presented with neonatal jaundice and diagnosed as GS by fragment analysis (A(TA)7TAA allele in homozygous state). He required periodical phenobarbitone therapy and was asymptomatic until puberty when his bilirubin levels significantly raised, and CN II was suspected. A bidirectional sequencing of five exons and exon/intron boundaries of the gene UGT1A1 (OMIM: 191,740) was performed. Four different variants in the UGT1A1 gene were identified in the patient: g.3664A > C (c.1352A > C, rs3755319); g.4963_4964TA [7] (c.-53_-52insTA, A(TA)7TAA, UGT 1A1*28, rs8175347); g.5884G > T (c.864 + 5G > T, IVS1 + 5G > T); and g.11895_11898del (c.996 + 2_996 + 5del). In the ClinVar database, the variant g.4963_4964TA [7] is described as a variant affecting response to drug treatment. This is the most common variant identified in patients with GS. The second intronic variant g.11895_11898del was reported for the first time [15]. In pediatric patients, Maruo et al [16] showed that the levels of serum bilirubin varied continuously within the spectrum from GS to CN II depending on genotypes. Serum bilirubin concentrations of typical CN II was 12.9 ± 5.1 mg/dl, the intermediate group was 5.2 ± 2.2 mg/dl, and typical GS was 2.8 ± 1.1 mg/dL (P < 0.0001) [16].
Drug therapy with phenobarbitone to reduce the levels of unconjugated bilirubin in CN II is well known; however, studies have reported that phenobarbital may cause hyperactivity, behavioral problems, sedation, and even dementia; these effects are dose related to some extent [17]. One of the reviews showed that phenobarbitone was significantly more likely to be associated with withdrawl than phenytoin in epileptic patients [18]. However, most of the studies with regard to safety profile of phenobarbitone are in the context of epileptic patients. There is hardly any data on the safety profile of phenobarbitone in patients with CN syndrome II, and data is lacking regarding the long-term neurodevelopmental outcome of those taking phenobarbitone.
Conclusion
Early suspicion of inherited causes of jaundice must prompt the evaluation of genetic mutations to look for coexistence of mutations for both GS and CN II. Coexistence of these mutations have implication on long-term neurological complications of unconjugated hyperbilirubinemia in stress or crisis situations. Prenatal diagnostic testing must be advised for detecting homozygous UGT1A1 mutations to diagnose CN II and Gilbert mutations for each of the future pregnancies. Although there are no clear guidelines as to when to stop phenobarbitone therapy in CN II patients, considering the side effects of long-term phenobarbitone therapy, the decision can be taken on case-to-case basis of stopping the therapy while monitoring TSB levels.
Patient perspective
The child’s father expresses the following: “We were quite anxious regarding the prolonged jaundice of our daughter in the early infancy. After the diagnosis was made by genetic tests, the presence of double mutations was scary; however, soon after the treatment was started (oral phenobarbitone syrup), she has been as normal as other children, growing well, is able to do all her regular activities, and is good at academics too; we wished to discontinue the medication soon and look for the recurrence of jaundice. We have been counseled regarding the recurrence of similar jaundice causing mutations in the next pregnancy and advised to undergo prenatal genetic diagnosis.”
Availability of data and materials
The data that support the case findings are available with the corresponding author on request.
Abbreviations
- CN II:
-
Crigler-Najjar syndrome type II
- CN I:
-
Crigler-Najjar syndrome type I
- GS:
-
Gilbert’s syndrome
- UDPGT:
-
Uridine diphospho-glucuronosyltransferase
- BIND:
-
Bilirubin-induced neurological damage
- NGS :
-
Next-generation sequencing
- TSB:
-
Total serum bilirubin
References
Stark AR (2020) Neonatal hyperbilirubinemia. In: Eichenwald EC et al (eds) Cloherty and Stark’s Manual of Neonatal Care, 9th edition Wolters Kluwer. pp 347–66
Crigler JF Jr, Najjar VA (1952) Congenital familial non-hemolytic jaundice with kernicterus. Paediatrics 10(2):169–180
Ebrahimi A, Rahim F (2018) Crigler-Najjar syndrome: current perspectives and the application of clinical genetics. Endocr Metab Immune Disorder Drug Targets 18(3):201–211
Skierka JM, Kotzer KE, Lagerstedt SA, O’Kane DJ, Baudhuin LM (2013) UGT1A1 genetic analysis as a diagnostic aid for individuals with unconjugated hyperbilirubinemia. J Pediatric 162:1146–1152
Van Es HHG et al (1993) Assignment of the human UDP-glucuronosyltransferase gene (UGT1A1) to the chromosome region 2q37. Cytogenetic Cell Genet 63:114–116
Alexander NM (2016) Metabolic diseases of the liver. In: Kliegman RM et al (eds) Nelson Textbook of Pediatrics, 20th edn. Elsevier, Philadelphia, pp 1937–38
Bosma P, Chowdhury JR, Jansen PH (1995) Genetic inheritance of Gilbert’s syndrome. Lancet 346:314–315
Abell R, Monteleone B, Chawla A (2012) Case of severe unconjugated hyperbilirubinemia in a neonate heterozygous for Gilbert syndrome. Hereditary Genet 1:114
Chalasani N et al (1997) Kernicterus in an adult who is heterozygous for Crigler-Najjar syndrome and homozygous for Gilbert-type genetic defect. Gastroenterology 112(6):2099–2103
Kadakol A et al (2001) Interaction of coding region mutations and the Gilbert-type promote abnormality of the UGT1A1 gene causes moderate degrees of unconjugated hyperbilirubinemia and may lead to neonatal kernicterus. J Med Genetics 38(4):244–249
Mane S, Ronghe A (2017) Gilbert syndrome: a case series. Pediatr Oncall J 14:41–2
Sood V, Lal BB, Sharma S et al (2021) Gilbert’s syndrome in children with unconjugated hyperbilirubinemia – an analysis of 170 cases. Indian J Pediatr 88:154–157
Cozzi L, Nuti F, Degrassi I, Civeriati D, Paolella G, Nebbia G (2022) Gilbert or Crigler-Najjar syndrome? Neonatal severe unconjugated hyperbilirubinemia with P364L UGT1A1 homozygosity. Ital J Pediatr 48(1):59
Sun L, Li M, Zhang L, Teng X, Chen X, Zhou X, Ma Z, Qi L, Wang P (2017) Differences in UGT1A1 gene mutations and pathological liver changes between Chinese patients with Gilbert syndrome and Crigler-Najjar syndrome type II. Medicine (Baltimore) 96(45):e8620
Gailite L, Rots D, Pukite I, Cernevska G, Kreile M (2018) Case report: Multiple UGT1A1 gene variants in a patient with Crigler-Najjar syndrome. BMC Pediatr 18(1):317
Maruo Y, Nakahara S, Yanagi T, Nomura A, Mimura Y, Matsui K, Sato H, Takeuchi Y (2016) Genotype of UGT1A1 and phenotype correlation between Crigler-Najjar syndrome type II and Gilbert syndrome. J Gastroenterol Hepatol 31(2):403–408
Iivanainen M, Savolainen H (1983) Side effects of phenobarbital and phenytoin during long-term treatment of epilepsy. Acta Neurol Scand Suppl 97:49–67
Nolan SJ, Smith CT, Pulman J, Marson AG (2013) Phenobarbitone versus phenytoin monotherapy for partial onset seizures and generalized onset tonic-clonic seizures. Cochrane Database Syst Rev 31(1):CD002217
Acknowledgements
None
Funding
None.
Author information
Authors and Affiliations
Contributions
RS and SS have made substantial contributions to the conception of the case report; RS and SS designed the work. SS and CTD contributed to the case follow-up and in collecting the case details. RS and SS contributed to the literature search and case analysis and drafted the work. SS and CTD substantially revised it, and RS, SS, and CTD have approved the submitted version and have agreed to be personally accountable for the author’s own contributions to the case report.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Written consent taken from the parent is available.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rangan, R.S., Shah, S. & Deshmukh, C.T. Coexistence of mutations of Gilbert’s syndrome and Crigler-Najjar syndrome in an infant with unconjugated hyperbilirubinemia—a case report. Egypt Pediatric Association Gaz 71, 49 (2023). https://doi.org/10.1186/s43054-023-00192-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43054-023-00192-9