
Abstract
Background
Hemolytic uremic syndrome (HUS) is characterized by microangiopathic hemolytic anemia (MAHA), thrombocytopenia, and acute kidney injury. The acute pancreatitis-associated HUS is a rare entity, and this case is one of the few pediatric cases reported.
Case presentation
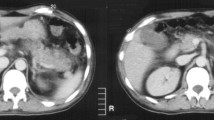
A 17-year-old girl referred to the emergency department with complaints of abdominal pain, fever, and vomiting. The skin and sclera were icteric. Murphy’s sign was positive. Hemogram was normal, biochemical analysis revealed an increase in liver function tests and cholestasis enzymes. Abdominal CT revealed multiple stones in the hydropic gallbladder lumen and the pancreas was edematous. With a diagnosis of acute pancreatitis supportive treatment was started. Acute kidney failure, cholestasis, anemia, and thrombocytopenia developed at the 2nd day of follow-up. Thrombotic thrombocytopenic purpura was excluded with normal ADAMTS-13 level. Intermittent hemodialysis and plasma exchange (PE) treatments were initiated, considering pancreatitis-related HUS. On the 3rd day of PE, the patient’s urine output exceeded 1 cc/kg per hour. No schistocyte was found in the peripheral smear after 7 PE and 5 hemodialysis sessions. Anemia, thrombocytopenia, and kidney functions improved. On the 15th day of the follow-up, endoscopic retrograde cholangiopancreatography performed, and gallbladder stones were removed.
Conclusion
The pathogenesis of HUS developing after acute pancreatitis is not fully understood. The mechanism proposed is that acute pancreatitis triggers cytokine release, resulting in endothelial damage and HUS. In conclusion, HUS may rarely develop in patients with acute pancreatitis. With the early diagnosis and appropriate treatment, the kidney functions can be completely normalized.
Similar content being viewed by others


Background
Hemolytic uremic syndrome (HUS) is characterized by microangiopathic hemolytic anemia (MAHA), thrombocytopenia and acute kidney injury. The most accepted classification in recent years is the etiology and physiopathology-based classification of HUS. While the term ‘typical HUS’ is used for STEC-HUS (Shiga toxin producing Escherichia coli-HUS), atypical HUS is grouped as primary atypical HUS (aHUS) caused by alternative complement pathway dysregulation and secondary aHUS. Secondary forms of aHUS can be listed as Streptococcus pneumoniae/Influenza A/H1N1-HUS, Cobalamin C defect-HUS, DGKE (diacylglycerol kinase ε) mutation-HUS, HUS with coexisting condition/disease (like transplantation/malignancy/drugs) [1]. The most common type in childhood is STEC-HUS, known as typical HUS, with a frequency of nearly 90% and should be excluded in the diagnosis of aHUS [2, 3]. HUS with acute pancreatitis as a coexisting condition, is a rarely reported secondary form of HUS. Aside from the ongoing debate on etiologic classification, the common end point of the disease is the inflammatory process that starts on the endothelial cell surface and the development of thrombotic microangiopathy with fibrin and platelet accumulation caused by this [3, 4]. The acute pancreatitis (AP)-related HUS mentioned in this article is a rare entity with nearly 30 cases reported in the literature, mostly from adult age. This case is also valuable as it is one of the few reported cases under the age of 18.
Case presentation
A 17-year-old Syrian girl presented to the emergency department with complaints of abdominal pain, fever, and vomiting. Her history revealed that she had a pregnancy history 1 year ago and had a healthy birth. In her physical examination, body weight was 96 kg (> 97p), height 165 cm (50-75p), blood pressure was 125/85 mmHg. The skin and sclera were icteric. She had abdominal tenderness and Murphy’s sign was positive. In laboratory examination, hemogram (hemoglobin 13.8 g/dL, platelet 218,000/μL) was within normal ranges. Biochemical analysis revealed an increase in liver function tests and cholestasis enzymes (ALT/AST 258/74 U/L, direct bilirubin 3.46 mg/dL, GGT: 232 U/L, amylase: 1387U/L, lipase 12558 U/L, creatinine 0.85 mg/dL). Urinalysis revealed a density of 1018 and there was 81 erythrocytes/HPF. Abdominal CT revealed multiple stones in the hydropic gallbladder lumen and the pancreas was edematous. With the preliminary diagnosis of AP, intravenous hydration and infusion of 3rd generation cephalosporin and octreotide were started.
The patient’s urine output decreased to 0.4 cc/kg per hour, who developed temporary vision loss after generalized grand-mal seizure at the 48th hour of the follow-up. Impairment in kidney function tests, increase in cholestasis enzymes (urea 102 mg/dL, creatinine 1.81 mg/dL, U/L, total bilirubin 45 mg/dL, direct bilirubin 26.4 mg/dL), anemia and thrombocytopenia (hemoglobin 9.5 g/dL, platelet 37,000/μL) developed. Coagulation parameters were in normal range. Schistocytes, fragmented erythrocytes polychromasia of the blood smear, accompanying low haptoglobin (< 8 mg/dL) and high LDH levels (2053 μ/L) were present. Direct Coombs was negative. C3 and C4 levels were 0.65 g/L (RR 0.79–1.5) and 0.14 g/L (RR 0.16–0.38), respectively. Thrombocytopenic purpura was excluded with normal ADAMTS-13 level. Creatinine rose to 2.98 mg/dL, anemia (6.6 g/dL) and thrombocytopenia (16,000/μL) became evident the day after. Since AP findings were present and there was no finding suggestive of hemolysis at hospital admission, intermittent hemodialysis (HD) and plasma exchange (PE) were initiated, considering AP-related HUS. The patient's urine output exceeded 1 cc/kg/h after the third PE (5th day of hospitalization). No schistocyte was found in the peripheral smear and complete hematological remission was achieved on the 11th day of hospitalization. After 7 sessions of PE (30 ml/kg) and 5 sessions of HD anemia, thrombocytopenia, and kidney functions (hemoglobin 10.1 g/dL, thrombocyte 204,000/μL, urea 31 mg/dL, creatinine 0.54 mg/dL) improved. In the second week of the follow-up, it was observed that the C3 and C4 values increased to 1.0 g/L and 0.2 g/L, respectively. It remained stable even after plasmapheresis was discontinued. However, the patient needed 2 more erythrocyte transfusion due to anemia. The changes in laboratory values during the follow-up are summarized in the Table 1.
On the 15th day of the follow-up, Endoscopic Retrograde Cholangiopancreatography performed, and gallbladder stones were removed, and a stent was placed in the common bile duct. On the 20th day of the patient's hospitalization, she had fever again. Pancreatic abscess developed as a complication of AP. She was discharged on the 42nd day of her hospitalization without sequelae.
Discussion
aHUS is a rare disease characterized by alternative complement pathway dysregulation which have multiple diverse etiologies. AP-related HUS mentioned in this article constitutes a small group among them [1]. A triggering event starts the inflammatory process on the endothelial cell surface. Fibrin and platelet deposition caused by endothelial cell damage evokes thrombotic microangiopathy and affects all organs and tissues, especially the kidneys [4]. Although the pathogenic mechanism of pancreatitis-related HUS has not been clarified yet, two basic pathologies have been suggested. The first is that the cytokines, interleukin-1, and tumor necrosis factor-α, released during AP directly cause vascular endothelial damage [5]. Another suggested triggering mechanism of AP-related HUS is it is the interaction of pancreatic proteases, which are released into the circulation with pancreatic tissue destruction with coagulation factors [5,6,7]. In healthy people, von Willebrand factor is degraded by ADAMTS13, preventing platelet aggregation [8]. Circulating proteases stimulate endothelial cell release of ultra large von Willebrand factor multimers and prevent the cleavage of these multimers by ADAMTS13, causing thrombus formation from shear stress on the vascular endothelium [5,6,7]. Considering this case and suggested pathogenic pathways, it can be speculated that the severity of AP and inflammation may be the triggering mechanism. However, no relationship was found between the severity of acute pancreatitis and the development of HUS in previous studies [9].
As in our case, development time of AP-related HUS is generally on the 3rd day of the onset of the disease and generally in period when pancreatitis is in resolution. This period between AP and HUS development provides evidence supporting these pathogenic pathways [10]. Due to the lack of available tests at our institution, we could not perform genetic tests to evaluate mutations in complementary regulatory proteins to rule out primary aHUS. However, the presence of still unidentified mutations makes it difficult to explain the etio-pathogenesis of the disease with the result of genetic mutation alone [11]. Sharma et al. in their article, suggested the existence of 3 types for the coexistence of AP and HUS, namely AP-associated secondary HUS, HUS triggered by AP, and TMA associated with HUS and AP. Accordingly, the most common type is AP-associated secondary HUS type of co-existence, had classical risk factors of AP like alcoholism or gall stones. It has been suggested that interleukins, TNF alpha and cytokines cause secondary HUS by causing endothelial damage [12]. Our case can be considered as type 1 coexistance due to the coexistence of gallstones and the age of development of HUS.
Pancreatitis-related HUS was first described in 1978 in an 18-year-old patient, and the etiology of pancreatitis was reported as idiopathic. After that, approximately 30 patients have been reported. The median age of these patients is 39 and the majority of them have alcoholic pancreatitis. To our knowledge, gallstone associated pancreatitis, as in our case, was reported in only four patients, and one of the patients had recurrent HUS [9]. Until now, there has been no reported patient under the age of 18, our case is the youngest age patient reported.
The presence of accompanying neurological symptoms was previously thought to distinguish between HUS and thrombotic thrombocytopenic purpura (TTP), which is a common cause of thrombotic microangiopathy, especially in the adult population. However, it is now known that central nervous system involvement is a common extrarenal complication of HUS especially in children [13]. A normal level of ADAMTS13 activity in a sample taken prior to initiation of plasma therapy safely excludes the diagnosis of thrombotic thrombocytopenic purpura [11]. In 2007, Swisher et al. reported a systemic review in patients with TTP-HUS. In this case series, 4 patients with neurological findings including diplopia, grand mal seizure, hemiparesis, and speech disorder were mentioned. ADAMTS-13 levels were not studied in any of these patients [5]. Although our case experienced temporary vision loss after generalized grand-mal seizure, normal ADAMTS13 activity and level results suggested that this neurological finding was an extrarenal manifestation of HUS in our patient.
Prior to the introduction of Eculizumab, which provides alternative complement system blockade via C5a blockade, plasma therapy was the mainstay of treatment in aHUS cases. Later there have been publications reporting that Eculizumab is much more favorable in this patient group due to its efficacy and low side-effect profile compared to plasma therapy [1, 14]. Due to the high cost of eculizumab and its low availability, especially in developing countries, a consensus guideline for developing countries was published in 2019. It is recommended early PE (not plasma infusion) therapy be continued at a daily rate until hematological remission achievement the absence of Eculizumab. Eculizumab treatment is recommended in the presence of unresponsiveness to PE therapy, life-threatening conditions, complications of PE, and hereditary complement defects [15]. However, there are no prospective, controlled studies to properly evaluate the efficacy of eculizumab in the treatment of secondary HUS cases such as pancreatitis-related HUS. In the reported cases of pancreatitis related HUS, it was observed that those who used eculizumab did not need dialysis therapy and a rapid remission of the disease was achieved [9]. Since our case was a Syrian refugee and had payment problems, we started PE as first-line treatment. Fortunately, the patient responded quickly and completely to PE.
Conclusion
The pathogenic mechanism of pancreatitis-related HUS, which has been reported with a small number of adult cases, is not fully understood. The mechanism proposed is that acute pancreatitis triggers cytokine release, resulting in endothelial damage and HUS. About half of the reported cases were seen after alcoholic pancreatitis, and gallstone-associated pancreatitis as in our case was reported in very few patients. Most of the patients benefited from PE therapy, and eculizumab therapy is a reasonable treatment option in selected cases that are resistant to plasma exchange therapy and recur, especially in developing countries.
In conclusion, HUS may rarely develop in the course of AP. With early diagnosis and appropriate treatment of this complication, the kidney functions can be completely normalized.
Availability of data and materials
Not applicable
Abbreviations
- HUS:
-
Hemolytic uremic syndrome
- MAHA:
-
Microangiopathic hemolytic anemia
- STEC-HUS:
-
Shiga toxin producing Escherichia coli-HUS
- aHUS:
-
Atypical HUS
- HD:
-
Hemodialysis
- PE:
-
Plasma exchange
- TTP:
-
Thrombotic thrombocytopenic purpura
References
Loirat C, Fakhouri F, Ariceta G, Besbas N, Bitzan M et al (2016) An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr Nephrol 31(1):15–39. https://doi.org/10.1007/s00467-015-3076-8
Khalid M, Andreoli S (2019) Extrarenal manifestations of the hemolytic uremic syndrome associated with Shiga toxin-producing Escherichia coli (STEC HUS). Pediatr Nephrol 34(12):2495–2507. https://doi.org/10.1007/s00467-018-4105-1
Walsh PR, Johnson S (2018) Treatment and management of children with haemolytic uraemic syndrome. Arch Dis Childhood. 103(3):285–291. https://doi.org/10.1136/archdischild-2016-311377
Brocklebank V, Wood KM, Kavanagh D (2018) Thrombotic micro-angiopathy and the kidney. Clin J Am Soc Nephrol 13:300–317. https://doi.org/10.2215/CJN.00620117
Swisher KK, Doan JT, Vesely SK, Kwaan HC, Kim B et al (2007) Pancreatitis preceding acute episodes of thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: report of five patients with a systematic review of published reports. Haematologica. 92(7):936–943. https://doi.org/10.3324/haematol.10963
Singh NP, Aggarwal NP, Shah HR, Jha LK, Kumar A (2017) Hemolytic-uremic syndrome complicating acute pancreatitis. Indian J Crit Care Med 21:534–536. https://doi.org/10.4103/ijccm.IJCCM_121_17
Bernardo A, Ball C, Nolasco L, Moake J, Dong JF (2004) Effects of inflammatory cytokines on the release and cleavage of the endothelial cell-derived ultra- large von Willebrand factor multimers under flow. Blood 104:100–106. https://doi.org/10.1182/blood-2004-01-0107
Mannucci PM, Peyvandi F (2007) TTP and ADAMTS13: when is testing appropriate? Hematol Am Soc Hematol Educ Prog 2007(1):121–126. https://doi.org/10.1182/asheducation-2007.1.121
Sandino-Pérez J, Gutiérrez E, Caravaca-Fontán F, Morales E, Aubert-Girbal L et al (2021) Haemolytic uraemic syndrome associated with pancreatitis: report of four cases and review of the literature. Clin Kidney J 14:1946–1952. https://doi.org/10.1093/ckj/sfaa245
Silva VA (1995) Thrombotic thrombocytopenic purpura/hemolytic uremic syndrome secondary to pancreatitis. Am J Hematol 50:53–56. https://doi.org/10.1002/ajh.2830500111
Taton O, Delhaye M, Stordeur P, Goodship T, Le Moine A et al (2016) An unusual case of haemolytic uraemic syndrome following endoscopic retrograde cholangiopancreatography rapidly improved with eculizumab. Acta Gastroenterol Belgica. 79:257–261 PMID: 27382949
Sharma H, Bhadauria D, Goel A, Yaccha M, Gurjar M et al (2022) Co-existence of acute pancreatitis with hemolytic uremic syndrome: "The dilemma of a rare organ cross-talk". Pancreatology. 22(6):823–825. https://doi.org/10.1016/j.pan.2022.06.255
Weil EL, Rabinstein AA (2021) Neurological manifestations of thrombotic microangiopathy syndromes in adult patients. J Thrombosis Thrombol 51:1163–1169. https://doi.org/10.1007/s11239-021-02431-5
Legendre CM, Licht C, Muus P et al (2013) Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. New Engl J Med 368:2169–2181. https://doi.org/10.1056/NEJMoa1208981
Bagga A, Khandelwal P, Mishra K, Thergaonkar R, Vasudevan A et al (2019) Hemolytic uremic syndrome in a developing country: consensus guidelines. Pediatric Nephrol 34:1465–1482. https://doi.org/10.1007/s00467-019
Acknowledgements
Not applicable
Funding
The authors declared that this study received no financial support.
Author information
Authors and Affiliations
Contributions
Concept: ST; Design: S.T. and I.E.T.K.; data collection and processing: I.E.T.K., İ.A; literature search: S.T., I.E.T.K; writing: S.T., I.E.T.K. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Written informed consent was obtained from the patient’s parents.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Taner, S., Karaçay, I.E.T. & Arslan, İ. Acute pancreatitis complicated by hemolytic uremic syndrome: a pediatric case. Egypt Pediatric Association Gaz 70, 43 (2022). https://doi.org/10.1186/s43054-022-00140-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43054-022-00140-z