
Abstract
Background
Endometrial cancer (EC) is a common malignant tumor in women with increasing mortality. The prognosis of EC is highly heterogeneous which needs more effective biomarkers for clinical decision. Here, we reported the effect of autophagy-related genes (ARGs) on the prognosis of EC.
Methods
The expression data of EC tissues and adjacent non-tumor samples were available from the TCGA dataset and 232 autophagy-related genes were from The Human Autophagy Database. A prognostic ARGs risk model was further constructed by using LASSO-Cox regression, and its prognostic and predictive value were evaluated by nomogram. Further functional analysis was conducted to reveal a significant signaling pathway.
Results
A total of 45 differentially expressed ARGs were obtained, including 18 upregulated and 27 downregulated genes. Eleven ARGs (BID, CAPN2, CDKN2A, DLC1, GRID2, IFNG, MYC, NRG3, P4HB, PTK6, and TP73) were finally selected to build ARGs risk. This signature could well distinguish between the high- and low-risk patients (survival analysis: P = 1.18E-10; AUC: 0.733 at 1 year, 0.795 at 3 years, and 0.823 at 5 years). Furthermore, a nomogram was plotting to predict the possibility of overall survival and suggested good value for clinical utility.
Conclusion
We established an eleven-ARG signature, which was probably effective in the prognostic prediction of patients with EC.
Similar content being viewed by others

Introduction
According to current report [1], endometrial cancer (EC) has kept rising in both incidence and mortality, despite advances in treatment methods which is still sixth rank cancer in women globally [2]. As the aging population and increasing of obesity, the EC incidence is expected to rise even further [3]. The classic treatment strategy for EC involves surgery and adjuvant therapy based on final pathology for early-stage disease. Early detection of EC at early stages has a relatively favorable prognosis [4]. The prognosis of advanced EC remains poor because of its high rates of recurrent, metastatic [5]. In addition, the median survival time is less than 12 months for advanced EC [6].
The current known molecular markers of EC include PTEN, PI3K/AKT/mTOR pathway, Ras/Raf pathway, HRD pathway, and other genetic architecture such as ARID1A, CTNNB1, FGFR2, HER2/neu, and p53 which has been performed diagnostic, prognostic, and/or predictive of response to EC [7].
Autophagy plays a crucial role in cellular physiology and is responsible for degrading dysfunctional organelles, intracellular microbes, and pathogenic proteins by lysosome which could defeat deficiencies that may lead to disease [8]. Meanwhile, autophagy has been reported involved in nutrient recycling and metabolic adaptation and could regulate and act as double-edged sword in the development of cancer. Mountains of research have exposed that autophagy involves in the physiological and pathophysiological processes of endometrium stroma cells and epithelial cells and is associated with the EC. Triggering or inhibiting the molecular markers of autophagy including p53, AMPK, and PI3K/AKT/mTOR pathways could influence the process of EC development. Some reports found ATG7, ULK4, and other genes of somatic mutations different expressions in the EC [9]. Additionally, autophagy has been linked to resistance to chemotherapy [10]. Though some studies discussed the association between autophagy genes and EC [11], the target genes of autophagy need to be deeply practiced in clinical application. Up to now, only a few work analyzed the link between autophagy genes and progression of EC in small data [12], which need to be verified by other methods. In this study, we obtained the expression data of EC tissues and adjacent non-tumor samples from The Cancer Genome Atlas (TCGA) public database and constructed a prognostic model with eleven autophagy-related genes (ARGs), which could accurately assess the prognostic risk of patients with endometrial cancer.
Method
Data acquisition
A total of 370 uterine corpus endometrial carcinoma (EC) tissues and 11 adjacent non-tumor samples were obtained from UCSC Xena (https://xenabrowser.net/), including mRNA expression matrix, clinical, and survival information. We downloaded 232autophagy-related genes (ARGs) from The Human Autophagy Database (http://www.autophagy.lu/index.html).
Clustering and differentially expressed ARGs analysis
The ARGs log-transformed values were used to perform tsne analysis using the R package “Rtsne” [13]. Then, “limma” package was used to identify the differentially expressed ARGs. Genes with fold change (FC) > 1 or < 0.5 and adjusted P-value < 0.05 were defined as differentially expressed genes [14].
Functional pathway analysis
The webtool Metascape (http://metascape.org/gp/index.html#/main/step1) was used to perform functional enrichment of differentially expressed ARGs. Gene Ontology (GO) biological processes, Reactome gene sets, Canonical pathways, Wiki Pathways, and the Kyoto Gene and Genomic Encyclopedia (KEGG) were used to assess relevant functional categories. Enrichment pathways with p and q values less than 0.05 are considered as significant categories [15].
Construction of prognostic signature based on ARGs
All DEGs were performed univariate Cox regression analysis to find out prognosis-related ARGs in EC. Then, significant genes (p-value < 0.05) were used to apply the least absolute shrinkage and selection operator (LASSO) Cox regression to narrow the range by using the “glmnet” R package [16]. The risk score formula was calculated as follows: risk score = (expressiongene1 × coefficientgene1) + (expressiongene2 × coefficientgene2) + … + (expressiongenen × coefficientgenen). Patients were classified into low-risk and high-risk groups based on the median value of risk scores. The Kaplan–Meier (K-M) method and time-dependent receiver operating characteristic (ROC) curve were generated to explore the prognostic accuracy of risk scores using the “survival” R package and “survivalROC” R package [17].
Establishment and assessment of the nomogram
For better clinical application of the risk signature, other clinicopathologic parameters, including age, tumor stage, neoplasm histologic grade, and histological type, were analyzed to explore the diagnostic capability of multigene prognostic signature. According to the results of multiple Cox regression, we construct a nomogram that can evaluate the OS probability of 1, 3, and 5 years by the “rms” package. The calibration plot and concordance index (C-index) analysis were performed to validate the nomogram.
Results
Identification of differentially expressed ARGs
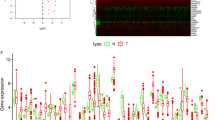
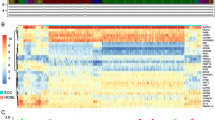
RNA-seq and clinical data from 370 EC tissue samples and 11 non-tumor samples were downloaded from TCGA, and ARGs were obtained from The Human Autophagy Database. As showed in Fig. 1A, ARGs appeared to have the potential ability to discriminate tumors from control samples. With criteria of |log2 FC|> 1 and adjusted P-value < 0.05, we finally obtained 45 differentially expressed ARGs, including 18 upregulated and 27 downregulated ARGs (Fig. 1B, C, Table S1). The pathway enrichment analysis indicated that above 45 genes participate in the autophagy pathway, apoptosis pathway, intrinsic apoptotic signaling pathway, and gastrin signaling pathway (Fig. 1D).

Identification of differentially expressed ARGs of EC. A Principal component analysis (PCA) between tumors and control samples; the result demonstrated the heterogeneity of the study. Differentially expressed ARGs in the study, 18 upregulated and 27 downregulated ARGs (|log2 FC|> 1, adjusted P-value < 0.05) was shown on B volcano plots and C hot map. Autophagy pathway, apoptosis pathway was major pathway by functional pathway analysis of ARGs was demonstrated on D
Construction of prognostic prediction model
A total of 11 ARGs were identified significantly associated with survival status using univariate Cox regression analysis (p < 0.05, Table 1). We then calculated the relative regression coefficient of 11 ARGs using LASSO Cox regression model, which screened out eleven ARGs in final, including BID (BH3 interacting domain death agonist), CAPN2 (calpain 2), CDKN2A (cyclin-dependent kinase inhibitor 2A), DLC1 (DLC1 Rho GTPase-activating protein), GRID2 (glutamate ionotropic receptor delta type subunit 2), IFNG (interferon gamma), MYC (MYC proto-oncogene, bHLH transcription factor), NRG3 (neuregulin 3), P4HB (prolyl 4-hydroxylase subunit beta), PTK6 (protein tyrosine kinase 6), and TP73 (tumor protein p73). The final risk scores were as followers: risk score = (0.4585 * expression value of BID) + (− 0.0999 * expression value of CAPN2) + (0.0357 * expression value of CDKN2A) + (− 0.0445 * expression value of DLC1) + (0.1136 * expression value of GRID2) + (− 0.0968 * expression value of IFNG) + (0.0758 * expression value of MYC) + (0.1947 * expression value of NRG3) + (− 0.3470 * expression value of P4HB) + (0.1536 * expression value of PTK6) + (− 0.1034 * expression value of TP73).
According to the median risk score in EC patients, 370 samples were divided into high-risk group (N = 185) and low-risk group (N = 185). K-M survival curves were used to analyze different survival times between high-risk and low-risk groups. The results showed that the high-risk group had a significantly poor prognosis (p = 1.18E − 10, Fig. 2A–D). Significantly, the area under the curve (AUC) of the corresponding receiver operating characteristic (ROC) curve for 1 year, 3 years, and 5 years of survival are 0.733, 0.795, and 0.823 respectively, which indicate that the risk score with11 ARGs has great ability in survival prediction of EC patients (Fig. 2E).

Construction of prognosis prediction model of EC. A Rank of risk score and distribution of groups. B The overall survival of patients in different groups. C Expression heatmap. D Kaplan–Meier survival analysis. E Time-dependent ROC curve analysis for survival prediction by the risk score
Independence of the risk score from other clinical parameters
In order to assess whether the risk score based on 11 ARGs is independent of other clinical variables including age, tumor stage, neoplasm histologic grade, and histological type, univariate and multivariate Cox regression analyses were performed. As showed in Fig. 3A, all clinical variables were significantly associated with the prognosis of EC (p < 0.05). Notably, the association between risk score and prognosis of EC patients remained after adjustment for four clinical values (p < 0.001, HR = 2.15, 95% CI = 1.58–2.92). Moreover, age (P = 0.049, HR = 1.97, 95% CI = 1.002–3.87), stage (P = 0.002, HR = 2.37, 95% CI = 1.37–4.1), and grade (P = 0.0496, HR = 1.97, 95% CI = 1.001–3.883) were also identified to be independent prognostic factors for EC (Fig. 3B).

Univariate (A) and multivariate (B) regression analyses of the prognostic value for EC
Construction of a nomogram for prediction 1-, 3-, and 5-year survival rate of EC
For the convenience of clinical application, a clinically quantitative method was expected to build up to predict the 1-, 3-, and 5-year survival rate of EC. Based on our results, we combined ARGs risk scores with three clinical values (age, stage, and grade) to produce a nomogram (Fig. 4A), and the C-index reached up to 0.816. In the calibration curve, the diagonal line (ideal model) represented the best prediction (Fig. 4B–D), which suggested that the nomogram had the fine prediction ability.

Construction of a nomogram for prediction survival of EC. A The nomogram to predict 1-, 3-, or 5-year survival rate. B–D The calibration plots for predicting patient 1-, 3-, or 5-year survival rate
Discussion
Autophagy, engulf and resolve cellular components for delivery to the lysosome, is the highly conserved process. Autophagy-related (ATG) genes belong to evolutionarily conserved genes and represent potential targets for oncotherapy. Current effort of inhibiting the lysosome by using chloroquine will be used to inhibit tumor cell growth or induce tumor cell death [18]. The association between endometrial cancer survival and autophagy proteins had been reported in many studies. Lebovitz et al. [9] had explored the association of 211 human autophagy-associated genes with tumor and found core autophagy mutation such as ATG4 C, ULK4, and RB1CC1/FIP200 significantly correlated with EC. Besides, high expression of FAM83B was associated with a poor prognosis in endometrial cancer [19]. Increased BECN1 expression was found related to poor estimated 5-year survival [20]. Conversely, CDKN2A, an autophagy activator highly expressed in non-endometrioid tumors [21]. Together, the previous evidence suggested that the autophagy gene may involve in EC development.
In this study, we profiled the mRNA expression of 232 autophagy-associated genes in the TCGA endometrial carcinoma cohort. The risk score was calculated for each patient by integrating mRNA expression levels and risk coefficients for selected genes. Among them, a total of 45 differentially expressed ARGs were obtained, and 11 ARGs (BID, CAPN2, CDKN2A, DLC1, GRID2, IFNG, MYC, NRG3, P4HB, PTK6, and TP73) were finally selected to build ARGs risk by using LASSO Cox regression model. Zhang et al. developed a four-gene (CDKN2A, PTK6, ERBB2, and BIRC5) prognostic signature for EC [12], suggesting their potential as independent predictive biomarkers and therapeutic targets for endometrial cancer. Since then, autophagy gene signature has been frequently performed to predict the prognosis of EC. When we compared our gene signature with Zhang’s, two genes (CDKN2A, PTK6) were in common between the two datasets, suggesting that the key autophagy associated genes of EC may impact by different stages and subtypes of EC.
Besides, plenty of clinical and basic research had investigated the role of ARGs in endometrial carcinoma. CAPN2 [22], GRID2 [23], IFNG [24], MYC [25], NRG3 [23], P4HB [26], PTK6 [12], and TP73 [27] was reported linked to endometrial carcinoma and dynein light chain 1(DLC1) contributed to cell cycle progression in estrogen-stimulated cells and affect the progression of breast and endometrial cancer [28]. Recently, several lines of evidence reported that targeting autophagy is a therapeutic approach in endometrial cancer. Bortezomib, a 26S proteasome inhibitor used to treat multiple myeloma had anticancer properties by inhibiting the NF-κB pathway [29]. Paclitaxel exposure in endometrial cancer cell lines will decrease p62 abundance [30].
Lastly, we developed a nomogram to predict individuals’ clinical outcomes. Nomogram is a useful tool to measure risk on an individual basis by combining and delineating risk factors, which has been used in endometrial carcinoma patients [31, 32]. A nomogram statistics risk factor (age, gender, stage) for the predictive model was offered in presenting graph. For expected to conduct that could predict the 1-, 3-, and 5-year survival rate of EC. We merge ARGs risk scores and three clinical values (age, stage, and grade) to produce a nomogram, and the C-index were 0.816 accurately. The combination group of the autophagy-gene signature and prognostic factors achieved better prognostic performance. Genomic and bioinformatics technologies have provided pioneering insight into the molecular etiology of endometrial tumors [33]. Moreover, according to molecular prognostic value of PORTEC-3 trial, sort the high-risk of EC. The recurrence-free survival with adjuvant chemotherapy and radiotherapy for p53 abnormal tumors was significantly higher compared to other subgroups, regardless of histologic type [34]. Despite the attempt for integrating gene markers and traditional risk factors in predicting the prognosis of EC, the usage in the clinical trial is striking limited. Briefly, we indicated that a nomogram, including an 11-autophagy gene signature, could well predict 1-, 3-, and 5-year survival possibilities of EC patients in the current study.
This study has several limitations. First, our results are based on a small sample size and retrospective analysis, which need confirmation in further cohorts. In addition, these findings were deduced from TCGA, which required clinical trials to practice. Furthermore, it needs additional biological experiments to demonstrate results.
Conclusion
We constructed a risk score with 11-autophagy related genes based on endometrial carcinoma of TCGA. And this risk score could independently predict the prognosis of EC patients. A nomogram combining gene signature and clinical risk factors could accurately predict the 1-, 3-, and 5-year survival probability for endometrial carcinoma patients. Our finding suggests that the 11-autophagy gene signature may help facilitate personalized medicine in the clinical setting.
Availability of data and materials
Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
- EC:
-
Endometrial carcinoma
- ARGs:
-
Autophagy-related genes
- KEGG:
-
Kyoto Gene and Genomic Encyclopedia
- LASSO:
-
Least absolute shrinkage and selection operator
- K-M:
-
Kaplan-Meier
- ROC:
-
Receiver operating characteristic
- C-index:
-
Concordance index
- BID:
-
BH3 interacting domain death agonist
- CAPN2:
-
Calpain 2
- CDKN2A:
-
Cyclin-dependent kinase inhibitor 2A
- DLC1:
-
DLC1 Rho GTPase-activating protein
- GRID2:
-
Glutamate ionotropic receptor delta type subunit 2
- IFNG:
-
Interferon gamma
- MYC:
-
MYC proto-oncogene
- bHLH:
-
Transcription factor
- NRG3:
-
Neuregulin 3
- P4HB:
-
Prolyl 4-hydroxylase subunit beta
- PTK6:
-
Protein tyrosine kinase 6
- TP73:
-
Tumor protein p73
References
Lu KH, Broaddus RR. Endometrial cancer. N Engl J Med. 2020;383(21):2053–64.
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
Isa T, Yamamoto E, Ozaki T, Kajiwara K, Morioka T. Use of pentazocine for anesthesia maintenance. Masui. 1970;19(13):1511–7.
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359-386.
Creutzberg CL, van Putten WL, Koper PC, Lybeert ML, Jobsen JJ, Warlam-Rodenhuis CC, De Winter KA, Lutgens LC, van den Bergh AC, van der Steen-Banasik E, Beerman H, van Lent M, Group PS. Survival after relapse in patients with endometrial cancer: results from a randomized trial. Gynecol Oncol. 2003 May;89(2):201–9.
Ray-Coquard I, Favier L, Weber B, Roemer-Becuwe C, Bougnoux P, Fabbro M, Floquet A, Joly F, Plantade A, Paraiso D, Pujade-Lauraine E. Everolimus as second- or third-line treatment of advanced endometrial cancer: ENDORAD, a phase II trial of GINECO. Br J Cancer. 2013;108(9):1771–7.
Arend RC, Jones BA, Martinez A, Goodfellow P. Endometrial cancer: molecular markers and management of advanced stage disease. Gynecol Oncol. 2018;150(3):569–80.
Mizushima N, Levine B. Autophagy in human diseases. N Engl J Med. 2020;383(16):1564–76.
Lebovitz CB, Robertson AG, Goya R, Jones SJ, Morin RD, Marra MA, Gorski SM. Cross-cancer profiling of molecular alterations within the human autophagy interaction network. Autophagy. 2015;11(9):1668–87.
Liu H, Zhang L, Zhang X, Cui Z. PI3K/AKT/mTOR pathway promotes progestin resistance in endometrial cancer cells by inhibition of autophagy. Onco Targets Ther. 2017;10:2865–71.
Eritja N, Chen BJ, Rodriguez-Barrueco R, Santacana M, Gatius S, Vidal A, Marti MD, Ponce J, Bergada L, Yeramian A, Encinas M, Ribera J, Reventos J, Boyd J, Villanueva A, Matias-Guiu X, Dolcet X, Llobet-Navas D. Autophagy orchestrates adaptive responses to targeted therapy in endometrial cancer. Autophagy. 2017;13(3):608–24.
Zhang J, Wang Z, Zhao R, An L, Zhou X, Zhao Y, Wang H. An integrated autophagy-related gene signature predicts prognosis in human endometrial cancer. BMC Cancer. 2020;20(1):1030.
Zhou H, Wang F, Tao P. t-Distributed stochastic neighbor embedding method with the least information loss for macromolecular simulations. J Chem Theory Comput. 2018;14(11):5499–510.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47.
Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, Chanda SK. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10(1):1523.
Tibshirani R. The lasso method for variable selection in the Cox model. Stat Med. 1997;16(4):385–95.
Heagerty PJ, Lumley T, Pepe MS. Time-dependent ROC curves for censored survival data and a diagnostic marker. Biometrics. 2000;56(2):337–44.
Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17(9):528–42.
Lin Q, Chen H, Zhang M, Xiong H, Jiang Q. Knocking down FAM83B inhibits endometrial cancer cell proliferation and metastasis by silencing the PI3K/AKT/mTOR pathway. Biomed Pharmacother. 2019;115:108939.
Giatromanolaki A, Koukourakis MI, Koutsopoulos A, Chloropoulou P, Liberis V, Sivridis E. High Beclin 1 expression defines a poor prognosis in endometrial adenocarcinomas. Gynecol Oncol. 2011;123(1):147–51.
Budina-Kolomets A, Hontz RD, Pimkina J, Murphy ME. A conserved domain in exon 2 coding for the human and murine ARF tumor suppressor protein is required for autophagy induction. Autophagy. 2013;9(10):1553–65.
Salehin D, Fromberg I, Haugk C, Dohmen B, Georg T, Bohle RM, Bauerschlag D, Maass N, Friedrich M. Immunhistochemical analysis for expression of calpain 1, calpain 2 and calpastatin in endometrial cancer. Anticancer Res. 2010;30(7):2837–43.
Chen X, Zhang W, Zhu H, Lin F. Development and validation of a 5-gene autophagy-based prognostic index in endometrial carcinoma. Med Sci Monit. 2021;12(27):e928949.
van Gool IC, Eggink FA, Freeman-Mills L, Stelloo E, Marchi E, de Bruyn M, Palles C, Nout RA, de Kroon CD, Osse EM, Klenerman P, Creutzberg CL, Tomlinson IP, Smit VT, Nijman HW, Bosse T, Church DN. POLE proofreading mutations elicit an antitumor immune response in endometrial cancer. Clin Cancer Res. 2015;21(14):3347–55.
Zhou WJ, Zhang J, Yang HL, Wu K, Xie F, Wu JN, Wang Y, Yao L, Zhuang Y, Xiang JD, Zhang AJ, He YY, Li MQ. Estrogen inhibits autophagy and promotes growth of endometrial cancer by promoting glutamine metabolism. Cell Commun Signal. 2019;17(1):99.
Colas E, Perez C, Cabrera S, Pedrola N, Monge M, Castellvi J, Eyzaguirre F, Gregorio J, Ruiz A, Llaurado M, Rigau M, Garcia M, Ertekin T, Montes M, Lopez-Lopez R, Carreras R, Xercavins J, Ortega A, Maes T, Rosell E, Doll A, Abal M, Reventos J, Gil-Moreno A. Molecular markers of endometrial carcinoma detected in uterine aspirates. Int J Cancer. 2011;129(10):2435–44.
Niwa Y, Hirose K, Matsuo K, Tajima K, Ikoma Y, Nakanishi T, Nawa A, Kuzuya K, Tamakoshi A, Hamajima N. Association of p73 G4C14-to-A4T14 polymorphism at exon 2 and p53 Arg72Pro polymorphism with the risk of endometrial cancer in Japanese subjects. Cancer Lett. 2005;219(2):183–90.
den Hollander P, Kumar R. Dynein light chain 1 contributes to cell cycle progression by increasing cyclin-dependent kinase 2 activity in estrogen-stimulated cells. Cancer Res. 2006;66(11):5941–9.
Di Lernia G, Leone P, Solimando AG, Buonavoglia A, Saltarella I, Ria R, Ditonno P, Silvestris N, Crudele L, Vacca A, Racanelli V. Bortezomib treatment modulates autophagy in multiple myeloma. J Clin Med. 2020;9(2):552.
Liu S, Li X. Autophagy inhibition enhances sensitivity of endometrial carcinoma cells to paclitaxel. Int J Oncol. 2015;46(6):2399–408.
Zhu L, Sun X, Bai W. Nomograms for predicting cancer-specific and overall survival among patients with endometrial carcinoma: a SEER based study. Front Oncol. 2020;10:269.
Luo Y, Mei D, Gong J, Zuo M, Guo X. Multiparametric MRI-based radiomics nomogram for predicting lymphovascular space invasion in endometrial carcinoma. J Magn Reson Imaging. 2020;52(4):1257–62.
Bell DW, Ellenson LH. Molecular genetics of endometrial carcinoma. Annu Rev Pathol. 2019;24(14):339–67.
Leon-Castillo A, de Boer SM, Powell ME, Mileshkin LR, Mackay HJ, Leary A, Nijman HW, Singh N, Pollock PM, Bessette P, Fyles A, Haie-Meder C, Smit V, Edmondson RJ, Putter H, Kitchener HC, Crosbie EJ, de Bruyn M, Nout RA, Horeweg N, Creutzberg CL, Bosse T, Trans PC. Molecular classification of the PORTEC-3 trial for high-risk endometrial cancer: impact on prognosis and benefit from adjuvant therapy. J Clin Oncol. 2020;38(29):3388–97.
Acknowledgements
The authors would like to thank TCGA for its free use. We also thank the Human Autophagy Database for providing great work.
Funding
This work was supported by Panzhihua Science and Technology Bureau (Nos.2020ZD-S-26), the Sichuan Medical Association Program (Nos.S20020).
Author information
Authors and Affiliations
Contributions
SyL and JP developed the study concept, designed, and interpreted the data, and YY Z drafted the manuscript. YT, XbZ, and ShW performed the data analysis. DxW, YyL, and DwX help specimen collection; JjL and DH supervised the design of the study and revised the manuscript. The authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethical approval and consent to participate
Ethical approval was obtained from the Ethics Committee of Panzhihua central hospital, 2021.12.15, pazszxyyll-2021–12.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, S., Pan, J., Zhang, Y. et al. An eleven autophagy-related genes-based prognostic signature for endometrial carcinoma. J Egypt Natl Canc Inst 34, 42 (2022). https://doi.org/10.1186/s43046-022-00135-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43046-022-00135-2