
Abstract
Hypertriglyceridemia (HTG) is a very common, yet underappreciated problem in clinical practice. Elevated triglyceride (TG) levels are independently associated with atherosclerotic cardiovascular disease (ASCVD) risk. Furthermore, severe HTG may lead to acute pancreatitis. Although LDL-guided statin therapy has improved ASCVD outcomes, residual risk remains. Recent trials have demonstrated that management of high TG levels, in patients already on statin therapy, reduces the rate of major vascular events. Few guidelines were issued, providing important recommendations for HTG management strategies. The goal of treatment is to reduce the risk of ASCVD and acute pancreatitis. The management stands on lifestyle modification, detection of secondary causes of HTG and pharmacological therapy, when indicated. In this guidance we review the causes and classification of HTG and summarize the current methods for risk estimation, diagnosis and treatment. The present guidance provides a focused update on the management of HTG, outlined in a simple user-friendly format, with an emphasis on the latest available data.
Similar content being viewed by others


Background
Hypertriglyceridemia (HTG) prevalence in adults varies between 10 and 29% according to the studied population [1]. HTG is classified as primary; due to genetic causes, or may be secondary to particular diseases, drugs or metabolic disorders [2].
Dyslipidemia itself usually causes no symptoms but can lead to symptomatic vascular disease, including coronary artery disease (CAD), cerebrovascular disease and peripheral arterial disease (PAD). In hereditary lipid disorders, a patient may present with the variants of cutaneous xanthoma. Eruptive xanthomas at pressure sites on the elbows, buttocks, and knees may be indicative of hypertriglyceridemia. High levels of TG (> 500 mg/dL) can cause acute pancreatitis. Higher TG levels can also cause dyspnea, hepatosplenomegaly lipemia retinalis and neurological manifestations such as confusion, transient memory loss, paresthesias and rarely, focal neurologic deficits. Extremely high lipid levels also give a lactescent (milky) appearance to blood plasma [3,4,5].
HTG is an underappreciated risk factor that is often overshadowed by low-density lipoprotein cholesterol (LDL-C) and other chronic conditions [6]. Studies have shown that despite the use of statin therapy, atherosclerotic cardiovascular disease (ASCVD) event rates remain high in patients with elevated TG [7]. The importance of lowering plasma levels of TG is currently considered an integral part of residual cardiovascular risk reduction strategies [8]. The American College of Cardiology/American Heart Association cholesterol guidelines list HTG as a risk-enhancing factor for cardiovascular disease (CVD) [9].
Dietary TGs are carried via the intestinal lymphatics in chylomicrons, while endogenous TGs produced by the liver are transported in very low density lipoproteins (VLDL) [10]. Large triglyceride-rich lipoproteins (TRL) particles such as nascent chylomicrons are unable to infiltrate the vessel wall [11]. However, cholesteryl-ester (CE)-enriched smaller TRLs (‘remnants’) can penetrate the subendothelium promoting atherogenesis through pro-inflammatory and pro-thrombotic pathways [12].
Therefore, ASCVD risk mediated by TRLs appears to be determined by the circulating concentration of apo B-containing particles rather than their TG content. An estimate for all apo B-containing lipoproteins is non-HDL-C [calculated as total cholesterol (TC) − HDL-C], hence its importance [13].
In addition, HTG is frequently associated with pathological high-density lipoprotein (HDL) particles that may add to the ASCVD risk [12].
Furthermore, HTG is believed to be causative in up to 10% of episodes of acute pancreatitis [14]. The risk of acute pancreatitis is a continuum with mild hypertriglyceridemia associated with a low long-term risk [15, 16], while more severe HTG is associated with progressively increased risk [16, 17].
We have previously published a consensus statement on the management of lipids [18]. We felt that there was a need to address an ignored and frequently overlooked part in the management of dyslipidemia which was hypertriglyceridemia. The following guidance is based on the best scientific evidence available till present and expert opinion considerations.
Scope of the article
The goal of this article is to provide practical guidance for clinicians in situations not covered by the current lipid guidelines, in a world with rapidly evolving evidence.
There are current gaps in care for high-risk patients with mild to moderate as well as severe forms of HTG. There are also limited data on management of patients at low to moderate atherosclerotic cardiovascular risk and moderately elevated triglycerides.
The evidence from clinical trials on triglyceride risk-based agents for ASCVD risk reduction is evolving and newer medications are soon coming into play. In this document, we aim to fulfill the following aspects:
-
1.
Definition and categories of HTG
-
2.
Role of lifestyle intervention in patients with HTG
-
3.
Role of statin therapy in patients with HTG
-
4.
Triglyceride risk-based non-statin therapies; when and how to give them?
-
5.
Providing practical algorithms for HTG management
Definitions and classifications
Hypertriglyceridemia is defined as a fasting plasma/serum TG level > 150 mg/dL or a non-fasting TG level > 175 mg/dL. We endorse the definition proposed by the American College of Cardiology on persistent hypertriglyceridemia; Fasting triglycerides ≥ 150 mg/dL following a minimum of 4–12 weeks of lifestyle intervention, a stable dose of maximally tolerated statin therapy when indicated, as well as evaluation and management of secondary causes of hypertriglyceridemia [7].
Patients with HTG can be categorized according to their fasting TG level as shown in Table 1.
Causes of hypertriglyceridemia
HTG may be either secondary to a variety of medical conditions or medications, or primary due to genetic causes [2]. Potential secondary causes for hypertriglyceridaemia [7] are listed in Table 2.
While secondary factors more frequently cause mild to moderate HTG, severe forms of HTG are more commonly primary in nature [10]. The causes of primary HTG are listed in Table 3 [19,20,21,22].
Who and when should we screen?
The age at which screening is recommended differs according to the level of risk as illustrated in Table 4.
Screening for HTG should also be considered in patients with [7]:
-
Acute pancreatitis
-
Suspected primary HTG:
-
Presence of tuberous xanthomas in the patient or family member
-
Family history of HTG
-
Severely elevated TG levels
-
Premature ASCVD in the patient or family member
-
-
Secondary causes for HTG
How should we screen?
For screening, lipid testing should include total cholesterol (TC), low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol (HDL-C), TG and non-HDL-C. It can generally be done using a non-fasting blood sample. The postprandial rise in TG is usually small, between 12 and 27 mg/dL. Fasting lipid testing is preferred in patients with metabolic syndrome, family history of premature ASCVD or genetic lipid disorders, non-fasting TG ≥ 400 mg/dL and to assess adherence to lifestyle interventions and lipid lowering medications [7, 9, 18, 23, 24].
Management of HTG in patients without ASCVD or diabetes mellitus (DM)
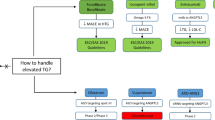
Limited data are available on ASCVD risk reduction using TG risk-based non-statin therapies for primary prevention in non-diabetic patients with persistent mild to moderate HTG [7]. Management of these patients is described in Fig. 1. However, patients with more severe forms of HTG have a relatively higher incidence of acute pancreatitis [7]. Management of these patients is described in Fig. 2.

Management of HTG in patients without ASCVD or DM. *ASCVD risk-enhancing factors including family history of premature ASCVD, persistently elevated LDL-C ≥ 160 mg/dL, chronic kidney disease, metabolic syndrome, inflammatory diseases (especially rheumatoid arthritis, psoriasis), biomarkers (persistently elevated fasting triglycerides ≥ 150 mg/dL, hs-CRP ≥ 2.0 mg/L) [7].

Management of HTG ≥ 500 mg/dl in patients without ASCVD or DM
Management of HTG in patients with ASCVD
Individuals with persistent moderate to severe HTG may be at increased risk of premature CVD. Few studies have shown an independent relationship, although small, between HTG and CVD. Studies have suggested that the ApoB-containing particles and/or lipoprotein (a), rather than the TG content of TRLs and their remnants are the cause of ASCVD risk [25, 26]. Management of HTG in ASCVD patients is illustrated in Fig. 3.

Management of HTG in patients with ASCVD
Management of HTG in patients with DM
The leading cause of mortality in diabetes mellitus is CVD. Diabetic patients have higher risk of developing ASCVD and much of this risk can be attributed to the development of atherogenic dyslipidemia, in which HTG is an essential component.
In well-controlled DM, the lipid profile may be normal, while in poorly controlled DM, the lipid profile shows a triad of HTG, increased levels of small dense LDL, and low HDL [27]. The management algorithm of HTG in diabetic patients is shown Figs. 2 and 4.

Management of HTG in diabetic patients without ASCVD
Management of HTG in patients with chronic kidney disease (CKD)
HTG is frequently encountered in the setting of CKD, especially in association with nephrotic syndrome and in patients on peritoneal dialysis [28]. The exact mechanisms through which CKD causes HTG are not exactly known, however, increased catabolism of VLDL [due to increased lipoprotein lipase (LPL) inhibitors as apo-C III] and increased VLDL production have been suggested [29]. Risk factors for development of HTG in CKD include DM, the degree of glomerular filtration rate (GFR) affection, severity of proteinuria, the modality of renal replacement therapy (peritoneal dialysis more than hemodialysis), nutritional status of the patient, and the presence of other comorbidities [14].
The aim of treatment is to primarily reduce the patient’s cardiovascular (CV) risk and in patients with severe HTG, reduce the risk of pancreatitis [14]. Non-pharmacological treatment is indicated as in patients without CKD, but diet modification is not recommended in malnourished patients. Statins are the cornerstone of treatment and are recommended to all patients with non-dialysis-dependent stage 3 or higher CKD, or recipients of renal transplantation. They should not be started in dialysis-dependent patients but should be continued if already on statins before being dialysis-dependent [23]. Caveats of statin use in CKD include the need for dose adjustment of some statins and the higher risk of rhabdomyolysis when used with cyclosporine or fibrates [11]. Icosapent Ethyl should be considered if HTG persists on statin therapy [30]. Whether there is a net benefit or not on CV outcomes with fibrate use in CKD is still unknown. Fibrates are therefore not recommended for the reduction of CV risk in CKD patients [14]. They may be considered in patients with severe HTG who are at high risk of pancreatitis under specialist consultation. Fibrates should be adjusted to GFR. Fibrates may lower cyclosporine levels in patients with renal transplants [14]. Lomitapide may be considered in patients with severe HTG who are at high risk of pancreatitis, with dose adjustment to GFR [31].
Management of HTG in patients with familial hypertriglyceridemia (FHTG)
There are five different types of FHTG classified according to the type of elevated lipoprotein and associated lipid changes including: familial combined hyperlipidemia (FCH), which is the commonest, familial chylomicronemia syndrome (FCS), familial dysbetalipoproteinaemia (FD), familial HTG (FHTG), and multifactorial chylomicronemia syndrome (MFCS) (Table 3) [19,20,21,22].
Pharmacologic management is frequently needed in FHTG to prevent pancreatitis and/or reduce risk of CVD. Medications commonly used for TG lowering are icosapent ethyl, fibrates, and Niacin [32]. In individuals who are unresponsive to treatment, plasmapheresis has been used.
There has been considerable interest in developing new TG lowering agents, especially for treatment of FHTG, using approaches targeting LPL and its regulators because of the central role they play in TRL metabolism. Volanesorsen is an antisense oligonucleotide that binds to apolipoprotein C3 (APOC3) mRNA preventing APOC3 translation. It has been shown to lower circulating APOC3 levels by 70–90% and TG levels by 56–86% in clinical trials involving both FCS and non-FCS patients with moderate to severe HTG. A reduction in the number of episodes of pancreatitis was also reported. The monoclonal antibody evinacumab that blocks the action of angiopoietin-like protein 3 (ANGPTL3) has been found to lower TG levels up to 80% in patients with mild to moderate HTG, as well as LDL-C levels [33, 34].
Lifestyle interventions
HTG is closely associated with a sedentary lifestyle and visceral adiposity. Weight loss is considered the most effective lifestyle intervention to lower TG levels; the effect of weight loss on lowering TGs is variable [35]. Five to ten percent reduction in body weight is associated with a 20% decrease in TG [34]. Higher-fat, lower carbohydrate diets have better effect on TG levels compared with lower-fat, higher carbohydrate diets [36]. The reduction in TG was greater for the very low carbohydrate regimens (in which carbohydrates represent < 10% of calories) [37]. A higher-protein (31% of caloric intake) versus a standard-protein (18% of caloric intake) weight-loss diet results in more weight loss and TG lowering [38].
There are different types of fasting, as shown in Table 5. Weight loss and greater TG lowering were noticed with alternate day fasting. A summary of nutrition recommendations for patients with HTG is shown in Table 6.
Alcohol is an important cause of elevated TG levels; patients with HTG are advised to abstain completely.
Physical activity and exercise reduce TG levels by 7–30%; the response is variable depending on the type, duration, and intensity of activity. In addition, smoking cessation should be advocated as a part of lifestyle intervention.
Pharmacological management [32]
Classes of medications commonly used in the management of HTG include omega-3 fatty acids, fibric acid derivatives and niacin. High doses of a strong statin (simvastatin, atorvastatin, rosuvastatin) also lower TG by up to 30%. The effect of different drugs on different lipid components is illustrated in Table 7.
Fibrates [39]
Fibrates are commonly used to treat HTG. They increase the activity of LPL through peroxisome proliferator activated receptor (PPAR) alpha receptor agonist action, which hydrolyzes TG in TRL.
Fibrates may be associated with a reversible increase in serum creatinine; therefore the dose should be lowered in patients with mild-to-moderate renal disease (about one third of the usual dose) and should be avoided in severe renal impairment. Risk for myopathy/ rhabdomyolysis increases with renal impairment or concurrent use of HMG-CoA reductase inhibitors. Rarely, severe anemia, leukopenia, thrombocytopenia may occur so periodic blood counts are recommended.
Omega 3 fatty acids [40]
Icosapent ethyl and docosahexaenoic acid (DHA) are the active constituents of the omega 3 fatty acids. Higher doses of icosapent ethyl (2–4 g) significantly reduce the levels of TGs. Omega 3 fatty acids reduce hepatic VLDL-TG synthesis and secretion. They also increase plasma LPL activity that in turn enhances TG clearance from circulating VLDL particles. Caution should be exercised in patients allergic to fish. Recurrent atrial fibrillation or flutter has been reported with administration of high doses. Omega 3 fatty acids may increase liver alanine transaminase (ALT), therefore it should be monitored. They may prolong bleeding time; therefore, caution is needed in patients at high bleeding risk.
Niacin [32]
Niacin reduces VLDL synthesis, and may increase chylomicron TG removal from plasma. Caution is used in patients with gout, DM, gall bladder disease, CVD, renal or hepatic impairment, if the patient is taking anticoagulants or HMG-CoA reductase inhibitors or if symptoms of myopathy occur (monitor creatine phosphokinase).
Statins [32]
Statins are competitive inhibitors of the activity of HMG-CoA reductase enzyme. This results in the reduction of the levels of LDL. Statins reduce TG possibly through upregulation of the VLDL uptake by hepatocytes, as well as a reduction in the production rate of VLDLs. Statin-associated muscle symptoms (SAMs) may occur in 10–15%. Liver function tests should be checked at baseline, and when clinically indicated.
Conclusions
Management algorithms proposed in this document for the management of hypertriglyceridemia are simple, cover the clinical situations most commonly observed in routine practice and are designed to improve the quality of care. In all patients with HTG, risk stratification is mandatory and an attempt should be made to identify a cause or association. All patients with HTG should have an assessment of their LDL-C and non-HDL-C levels. The goal of therapy is to reduce ASCVD risk, as well as risk of pancreatitis. The first step of treatment involves implementing lifestyle changes and management of secondary causes, if present. Proper control of other ASCVD risk factors is recommended. Statins are advised as first line agent for ASCVD risk reduction. For patients with persistently elevated TG levels after non-pharmacologic interventions and despite maximally tolerated LDL-guided therapy, we suggest introducing additional drug therapy when indicated. Pharmacological management in persistent, as well as severe HTG, is centered on omega-3 fatty acid therapy and fenofibrate.
Future directions
HTG and its impact on ASCVD remain an under-explored frontier in lipidology. Research is needed to uncover the molecular basis and the dynamics of the different atherogenic particles in this disease process.
A risk stratification algorithm for acute pancreatitis in patients with severe HTG is needed.
More RCT-based evidence is needed for the use of fibrates to reduce CV risk in various subgroups e.g.: CKD patients and the elderly.
More data regarding the efficacy of IPE in reducing CV risk without concomitant statin use (e.g.: in patients intolerant to statins) is needed.
Further safety and efficacy data on novel drugs acting on existing pharmacological targets (e.g.: the novel selective PPAR alpha modulator-pemafibrate), or novel pharmacological targets (e.g.: apoC-III and ANGPTL-3) is needed.
Availability of data and materials
The dataset supporting the results and conclusions of this article will be available from the corresponding author on request.
Abbreviations
- ACC:
-
American College of Cardiology
- ANGPTL3:
-
Angiopoietin-like protein 3
- ApoB:
-
Apolipoprotein-B
- ApoC:
-
Apolipoprotein-C
- ASCVD:
-
Atherosclerotic cardiovascular disease
- CAD:
-
Coronary artery disease
- CE:
-
Cholesteryl-ester
- CKD:
-
Chronic kidney disease
- CV:
-
Cardiovascular
- CVD:
-
Cardiovascular disease
- CYP2CA:
-
Cytochrome-P 2CA
- DGAT:
-
Diacylglycerol acyltransferase
- DM:
-
Diabetes mellitus
- FCH:
-
Familial combined hyperlipidemia
- FCS:
-
Familial chylomicronemia syndrome
- FD:
-
Familial dyslipoproteinemia
- FHTG:
-
Familial hypertriglyceridemia
- GFR:
-
Glomerular filtration rate
- GPIHBP1:
-
Glycosylphosphatidylinositol-anchored high density lipoprotein binding protein-1
- HDL-C:
-
High density lipoprotein-cholesterol
- HIV:
-
Human immunodeficiency virus
- HMG-CoA:
-
Hydroxymethylglutaryl coenzyme-A
- Hs-CRP:
-
Highly-sensitive C-reactive protein
- HTG:
-
Hypertriglyceridemia
- IDL-C:
-
Intermediate density lipoprotein-cholesterol
- IPE:
-
Icosapent ethyl
- LDL-C:
-
Low density lipoprotein-cholesterol
- LMF1:
-
Lipase maturation factor 1
- LPL:
-
Lipoprotein lipase
- MCT:
-
Medium-chain triglyceride
- MFCS:
-
Multifactorial chylomicronemia syndrome
- mRNA:
-
Messenger ribonucleic acid
- PPAR:
-
Peroxisome proliferator activated receptor
- PTT:
-
Partial thromboplastin time
- SAM:
-
Statin-associated muscle symptom
- SPPARMa:
-
Selective PPAR alpha modulator
- T1DM:
-
Type-1 diabetes mellitus
- T2DM:
-
Type-2 diabetes mellitus
- TC:
-
Total cholesterol
- TG:
-
Triglyceride
- TRL:
-
Triglyceride-rich lipoprotein
- VLDL-C:
-
Very low density lipoprotein-cholesterol
References
Karpov Y, Khomitskaya Y (2015) PROMETHEUS: an observational, cross-sectional, retrospective study of hypertriglyceridemia in Russia. Cardiovasc Diabetol 14(1):1–14. https://doi.org/10.1186/S12933-015-0268-2
Chait A, Subramanian S (2000) Hypertriglyceridemia: pathophysiology, role of genetics, consequences, and treatment
Dyslipidemia—endocrine and metabolic disorders—MSD manual professional edition. https://www.msdmanuals.com/professional/endocrine-and-metabolic-disorders/lipid-disorders/dyslipidemia. Accessed 14 Nov 2021
Goyal A, Cusick AS, Bansal P. Familial Hypertriglyceridemia. StatPearls. Published online July 4, 2021. https://www.ncbi.nlm.nih.gov/books/NBK556024/. Accessed 14 Nov 2021
Nayak KR, Daly RG (2004) Images in clinical medicine. Eruptive xanthomas associated with hypertriglyceridemia and new-onset diabetes mellitus. N Engl J Med 350(12):1235–1235. https://doi.org/10.1056/NEJMICM030676
Elkhal I, Warden BA (2021) Clinical considerations for the management of hypertriglyceridemia. Am Fam Phys 103(6):325–326
Virani SS, Morris PB, Agarwala A et al (2021) 2021 ACC expert consensus decision pathway on the management of ASCVD risk reduction in patients with persistent hypertriglyceridemia. J Am Coll Cardiol. https://doi.org/10.1016/J.JACC.2021.06.011
Vrablík M, Češka R (2015) Treatment of hypertriglyceridemia: a review of current options. Physiol Res 64(Suppl 3):S331–S340. https://doi.org/10.33549/PHYSIOLRES.933199
Grundy SM, Stone NJ, Bailey AL et al (2019) 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol 73(24):3168–3209. https://doi.org/10.1016/j.jacc.2018.11.002
Laufs U, Parhofer KG, Ginsberg HN, Hegele RA (2020) Clinical review on triglycerides. Eur Heart J 41(1):99–109c. https://doi.org/10.1093/eurheartj/ehz785
Nordestgaard BG, Stender S, Kjeldsen K (1988) Reduced atherogenesis in cholesterol-fed diabetic rabbits. Giant lipoproteins do not enter the arterial wall. Arteriosclerosis. https://doi.org/10.1161/01.atv.8.4.421
Hegele RA, Ginsberg HN, Chapman MJ et al (2014) The polygenic nature of hypertriglyceridaemia: implications for definition, diagnosis, and management. Lancet Diabetes Endocrinol. https://doi.org/10.1016/S2213-8587(13)70191-8
Ference BA, Kastelein JJP, Ray KK et al (2019) Association of triglyceride-lowering LPL variants and LDL-C-lowering LDLR variants with risk of coronary heart disease. JAMA J Am Med Assoc. https://doi.org/10.1001/jama.2018.20045
Tonelli MA, Wanner C, Cass A et al (2013) Kidney Disease: Improving Global Outcomes (KDIGO) lipid work group. KDIGO clinical practice guideline for lipid management in chronic kidney disease. Kidney Int Suppl 3(3):1–315
Yang AL, McNabb-Baltar J (2020) Hypertriglyceridemia and acute pancreatitis. Pancreatology. https://doi.org/10.1016/j.pan.2020.06.005
Hypertriglyceridemia—UpToDate. https://www.uptodate.com/contents/hypertriglyceridemia. Accessed 20 Aug 2021.
Scherer J, Singh VP, Pitchumoni CS, Yadav D (2014) Issues in hypertriglyceridemic pancreatitis: an update. J Clin Gastroenterol. https://doi.org/10.1097/01.mcg.0000436438.60145.5a
Taha HSED, Badran HM, Kandil H et al (2021) Egyptian practical guidance in lipid management 2020. Egypt Hear J. https://doi.org/10.1186/s43044-021-00140-1
Dron JS, Hegele RA (2020) Genetics of hypertriglyceridemia. Front Endocrinol (Lausanne). https://doi.org/10.3389/FENDO.2020.00455
Chyzhyk V, Brown AS (2020) Familial chylomicronemia syndrome: a rare but devastating autosomal recessive disorder characterized by refractory hypertriglyceridemia and recurrent pancreatitis. Trends Cardiovasc Med. https://doi.org/10.1016/j.tcm.2019.03.001
Shah AS, Wilson DP (2015) Primary hypertriglyceridemia in children and adolescents. J Clin Lipidol. https://doi.org/10.1016/j.jacl.2015.04.004
Hassing HC, Surendran RP, Mooij HL, Stroes ES, Nieuwdorp M, Dallinga-Thie GM (2012) Pathophysiology of hypertriglyceridemia. Biochim Biophys Acta Mol Cell Biol Lipids. https://doi.org/10.1016/j.bbalip.2011.11.010
Mach F, Baigent C, Catapano AL et al (2020) 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J 41(1):111–188. https://doi.org/10.1093/eurheartj/ehz455
Langsted A, Freiberg JJ, Nordestgaard BG (2008) Fasting and nonfasting lipid levels influence of normal food intake on lipids, lipoproteins, apolipoproteins, and cardiovascular risk prediction. Circulation. https://doi.org/10.1161/CIRCULATIONAHA.108.804146
Khetarpal SA, Rader DJ (2015) Triglyceride-rich lipoproteins and coronary artery disease risk: new insights from human genetics. Arterioscler Thromb Vasc Biol. https://doi.org/10.1161/ATVBAHA.114.305172
Wang H, Eckel RH (2009) Lipoprotein lipase: from gene to obesity. Am J Physiol Endocrinol Metab. https://doi.org/10.1152/ajpendo.90920.2008
Hartz JC, de Ferranti S, Gidding S (2018) Hypertriglyceridemia in diabetes mellitus: implications for pediatric care. J Endocr Soc. https://doi.org/10.1210/js.2018-00079
Kwan BCH, Kronenberg F, Beddhu S, Cheung AK (2007) Lipoprotein metabolism and lipid management in chronic kidney disease. J Am Soc Nephrol. https://doi.org/10.1681/ASN.2006091006
Dušejovská M, Vecka M, Rychlík I, Žák A (2020) Dyslipidemia in patients with chronic kidney disease: etiology and management. Vnitr Lek. https://doi.org/10.36290/vnl.2020.082
Vijayaraghavan K, Szerlip HM, Ballantyne CM et al (2019) Icosapent ethyl reduces atherogenic markers in high-risk statin-treated patients with stage 3 chronic kidney disease and high triglycerides. Postgrad Med. https://doi.org/10.1080/00325481.2019.1643633
Alonso R, Cuevas A, Mata P (2019) Lomitapide: a review of its clinical use, efficacy, and tolerability. Core Evid. https://doi.org/10.2147/ce.s174169
Davidson MH, Toth PP (2004) Comparative effects of lipid-lowering therapies. Prog Cardiovasc Dis. https://doi.org/10.1016/j.pcad.2004.04.007
Cruz-Bautista I, Huerta-Chagoya A, Moreno-Macías H et al (2021) Familial hypertriglyceridemia: an entity with distinguishable features from other causes of hypertriglyceridemia. Lipids Health Dis. https://doi.org/10.1186/s12944-021-01436-6
Goldberg RB, Chait A (2020) A comprehensive update on the chylomicronemia syndrome. Front Endocrinol (Lausanne). https://doi.org/10.3389/fendo.2020.593931
Byrne A, Makadia S, Sutherland A, Miller M (2017) Optimizing non-pharmacologic management of hypertriglyceridemia. Arch Med Res. https://doi.org/10.1016/j.arcmed.2017.11.017
Jensen MD, Ryan DH, Apovian CM et al (2013) AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of cardiology/American Heart Association task force on practice guidelines and the obesity society. Circulation. https://doi.org/10.1161/01.cir.0000437739.71477.ee
Fechner E, Smeets ETHC, Schrauwen P, Mensink RP (2020) The effects of different degrees of carbohydrate restriction and carbohydrate replacement on cardiometabolic risk markers in humans—a systematic review and meta-analysis. Nutrients. https://doi.org/10.3390/nu12040991
Wycherley TP, Moran LJ, Clifton PM, Noakes M, Brinkworth GD (2012) Effects of energy-restricted high-protein, low-fat compared with standard-protein, low-fat diets: a meta-analysis of randomized controlled trials. Am J Clin Nutr. https://doi.org/10.3945/ajcn.112.044321
Elam MB, Ginsberg HN, Lovato LC et al (2017) Association of fenofibrate therapy with long-term cardiovascular risk in statin-treated patients with type 2 diabetes. JAMA Cardiol. https://doi.org/10.1001/jamacardio.2016.4828
Picard F, Bhatt DL, Ducrocq G et al (2019) Generalizability of the REDUCE-IT trial in patients with stable coronary artery disease. J Am Coll Cardiol. https://doi.org/10.1016/j.jacc.2019.01.016
Acknowledgements
None.
Funding
None.
Author information
Authors and Affiliations
Contributions
HT: main author, put the idea behind this review and wrote, revised and edited the manuscript. HK, NF, AO, MS, FF, HM, JB, MS, ME, MA, MM: contributed to the writing of this manuscript and has read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This research involved human subjects and was performed in accordance with the Declaration of Helsinki and approved by Cairo University Ethical Committee with reference number–Egypt.
Consent for publication
Not applicable.
Competing interests
No conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Taha, H.S.E.D., Kandil, H., Farag, N. et al. Egyptian practical guidance in hypertriglyceridemia management 2021. Egypt Heart J 73, 107 (2021). https://doi.org/10.1186/s43044-021-00235-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43044-021-00235-9