
Abstract
Background
Alpha synuclein (α-synuclein) is coded by SNCA gene and found in a helical form with phospholipids or in an unfolded arrangement in the cytosol and belongs to the synuclein family other than beta synuclein and gamma synuclein. It is a short protein made of 140 amino acids with three domains: an N-terminal lipid binding helix, a non-amyloid-ß component (NAC), and an acidic tail at the C-terminus. α-Synuclein is present in aggregated and fibrillar form in Lewy bodies and its association has been related to multiple system atrophy (MSA), Parkinson’s disease (PD), and Dementia with Lewy bodies (DLB). Our objective is to investigate and prioritise the possible nsSNPs in the α-synuclein protein that have been potentially connected to human neurodegenerative diseases.
Results
We used the series of computational tools to predict the mutation's harmful effect on three-dimensional structure of α-synuclein based on consensus approach. Our findings pointed to a significant computational blueprint for discovering nsSNPs connected to neurodegenerative illnesses from a large SNP data set while also minimising the expenses of experimentally showing harmful nsSNPs.
Conclusions
The prioritised G25S (rs1433622151), V66E (rs1261243630), and V77D (rs745815563) mutations can be employed in additional experimental studies to assess the α-synuclein protein mutation in relation to neurodegenerative illnesses and develop a therapeutics against them.
Graphical Abstract

Similar content being viewed by others


Background
Alpha synuclein (α-synuclein) is a protein that belongs to the synuclein family other than beta synuclein (β-Synuclein) and gamma synuclein (γ-Synuclein) [1]. Synucleins are soluble, unfolded neural proteins found only in vertebrates [2]. In solution, α-synuclein is a IDP (intrinsically disordered protein), suggesting that it does not have a single stable 3D structure [3]. α-Synuclein can be seen in a helical form with phospholipids or in an unfolded arrangement in the cytosol, implying that its dynamic nature that allows it to play distinct roles in different cellular locations. α-Synuclein is comprised of 140 amino acid residues and is coded by SNCA gene. It is a short protein which is acidic in nature having three domains: a N-terminal lipid binding helix, a non-amyloid-β component (NAC), and an acidic tail at the C-terminal. N-terminal region starts from amino residues 1 to 60, is an amphipathic in nature, and is marked by seven 11-residue repeats (XKTKEGVXXXX) that include a highly conserved KTKEGV. This region shows similarity to the helical region of apolipoproteins. Furthermore, these repeating sequences are linked to α-synuclein's ability to interact with lipid bilayers [4]. The central NAC region (residues 61 to 95) is hydrophobic in nature which is involved in aggregation of proteins. Within the synuclein family, only α-synuclein has this domain [5]. The C-terminal (residues 96 to 140) domain is an extremely acidic, proline-rich site with no definite structural tendency. This region is important for function and solubility of α-synuclein as well as for its interaction among different proteins [6]. Alpha synuclein is implicated in the regulation of synaptic vesicle dynamics, including vesicle clustering, mobilisation, and recycling. It interacts with synaptic vesicle-associated proteins such as synaptobrevin and synapsin, suggesting a role in modulating neurotransmitter release and synaptic transmission. Aggregated forms of alpha synuclein, such as oligomers and fibrils, have been shown to directly bind to TLR2 on microglia, astrocytes, and other immune cells in the brain. This interaction triggers TLR2 signalling and downstream activation of nuclear factor kappa B (NF-κB) and mitogen-activated protein kinase (MAPK) pathways, leading to the production of pro-inflammatory cytokines, chemokines, and reactive oxygen species (ROS). Chronic activation of the TLR2 pathway by alpha-synuclein aggregates contributes to the progressive neurodegeneration observed in PD and related synucleinopathies. Persistent neuroinflammation, oxidative stress, and dysregulation of immune responses driven by TLR2 activation can lead to synaptic dysfunction, neuronal loss, and ultimately, disease progression [47].
α-Synuclein is primarily present in the axon terminals of presynaptic neurons in the brain. It interacts with proteins and phospholipids in these terminals. It controls synaptic vesicle trafficking and associated neurotransmitter release [7,8,9]. It is present in large concentrations in the brain (hippocampus, neocortex, thalamus, substantia nigra, and cerebellum), with lower amounts in the non-neuronal glial cells, heart, muscle, and other tissues [10]. α-Synuclein is present in aggregated and fibrillar form in Lewy bodies [4]. Lewy bodies are the indicator of damaged brain neurons of individuals with Parkinson’s disease (PD) and Dementia [11]. Association of α-synuclein has been related to various conditions, including pure autonomic failure, multiple system atrophy (MSA), PD, and DLB [12]. α-Synuclein accumulation is a critical factor related with lysosome and proteasome reduction, it also supports the theory that cell degeneration in PD is caused by lysosomal and proteasomal dysfunction, poor protein clearance and protein aggregation, all of which lead to cell death [13, 49].
Some autosomal-dominantly hereditary familial PD lineages were found to have a point genetic mutation coding the presynaptic α-synuclein protein [11, 48]. The pathological similarity between the A30P patient and Idiopathic Parkinson’s disease (IPD) clearly support the hypothesis that familial PD induced by the mutation (A30P) in the gene of α-synuclein is not only clinically but also pathologically connected to IPD [14].
SNPs (single-nucleotide polymorphisms) are most commonly found DNA sequence variations which arise when a single nucleotide in the genome sequence is changed. Approximately 2% of all identified single-nucleotide polymorphisms that are linked to a variety of diseases are non-synonymous SNPs (nsSNPs) in protein-coding regions [15]. Deleterious SNP prediction aims to find out if an nsSNP will affect a protein's function and so lead to hereditary disease. Since SNPs can produce amino acid variations, which cause alterations in the stability of mRNA transcripts and also in the binding affinity of transcription factors, doing a comprehensive analysis of SNPs in the biological system is complex. In this context, in silico screening is a useful technique for researching SNPs [16]. In order to determine whether alterations are detrimental or not, computational methods developed, to forecast the effects of amino acid residue changes in proteins, act like an initial filter of potentially deleterious changes.
Our aim is to explore nsSNPs within the SNCA gene encoding α-synuclein, with a focus on their relevance to detecting neurological disorders. Employing an array of computational tools with consensus based approach, we delve into the analysis of missense SNPs in α-synuclein using diverse prediction algorithms. Subsequently, our investigation extends to molecular dynamics simulations aimed at evaluating the structural implications of potentially deleterious mutations. We hypothesise that discerning these variants could significantly influence the stability and conformational integrity of the protein, shedding light on mechanisms underlying neurological disorders.
Methods
Data sets
Amino acid sequence of α-synuclein was retrieved from UniProt database (https://www.uniprot.org) [17] having Accession No. AAA16117.1. Ensembl genome browser (https://asia.ensembl.org/index.html) [18] was used to collect the SNP data for α-synuclein coded SNCA gene.
Protein sequence analysis
The computational methods used in primary protein sequence analysis of protein produce results depending on a number of parameters. The protein sequence of α-synuclein was analysed using various in silico tools. ProtParam (https:// web.expasy.org/protparam) [19] was used to compute the physiochemical properties of α-synuclein including molecular mass, theoretical pI, composition of amino acid, estimated half-life, instability index, aliphatic index, and grand average of hydropathicity (GRAVY). SMART (http://smart.embl-heidelberg.de) [20] was used to predict functional domains present in α-synuclein. NetPhos 3.1 ([21, 22] was used to detect phosphorylation sites for threonine, tyrosine, and serine amino acid residues and GPS 3.0 (http://gps.biocuckoo.org/online.php) [23] was used to identify protein sequences for kinase-specific phosphorylation sites. TargetP 2.0 (https://services.healthtech.dtu.dk/service.php?TargetP-2.0) [24] was used to predict subcellular localisation of α-synuclein. Then, SignalP 5.0 (https://services.healthtech.dtu.dk/service.php?SignalP-5.0) [24] was used to predict signal peptides including their cleavage site. The TOPCONS (https://topcons.cbr.su.se) [25] was used to predict transmembrane domains and their positions and finally STRING (https://string-db.org) [26] was used to predict possible physical and functional interacting partners of α-synuclein.
Binding pocket analysis
The possible biding pockets of α-synuclein was predicted by CASTp (http://sts.bioe.uic.edu/castp/calculation.html) [27] which is based on the current theoretical and algorithmic insights in computational geometry.
Deleterious nsSNPs prediction in coding region
The algorithmic tools used to anticipate potentially damaging nsSNPs produce results based on their own mechanism of action to assess the nature (neutral or deleterious) of nsSNP which may alter the structure and function of a protein, perhaps contributing to disease. Various in silico tools were used to predict nsSNPs in α-synuclein to study their effect on structure and function of it.
I-Mutant 3.0
I-Mutant 3.0 (http://gpcr2.biocomp.unibo.it/cgi/predictors/I-Mutant3.0/I-Mutant3.0.cgi) [28] helps to assess changes in stability of protein depending on single-site variation in the protein sequence or structure. It uses an SVM (support vector machine)-based approach to analyse the alterations in Gibbs free energy (ΔG) as ΔΔG. Protein stability is reduced when the ΔΔG prediction value is zero, and enhanced when the ΔΔG prediction value is greater than zero. To estimate protein stability following a point mutation, we employed both sequence and structure of protein as input sources.
PolyPhen-2 (Polymorphism Phenotyping v2)
PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2) [29] evaluates the effect on structural integrity and functionality of a protein by an amino acid variation, based on basic physicochemical and statistical parameters. PolyPhen-2 value ranges from 0.0 to 1.0. Variants are classified as benign, possibly damaging, or probably damaging as the value rises from 0.0 to 1.0. Protein sequence was employed as an input resource.
PhD‐SNP (Predictor of human Deleterious Single-Nucleotide Polymorphisms)
PhD-SNP (https://snps.biofold.org/phd-snp/phd-snp.html) [28] is SVM-dependent technique for predicting if a single-site polymorphism in a protein is disease causing or not. If the output score is greater than 0.5, the mutant variation is identified as disease causing; otherwise, it is termed neutral.
PANTHER (Protein ANalysis THrough Evolutionary Relationships)
PANTHER (http://www.pantherdb.org/tools/csnpScoreForm.jsp) [30] is an online tool that classifies proteins/genes to assist in rapid analysis. It facilitates in the prediction of tracing back evolutionary preservation. If the result is probably benign, the preservation time would be less than 200 my, and the mutation is less likely to impact protein function. When the preservation time is between 200 and 450 my, the variant is possibly damaging, and the variation may alter protein function. When the preservation time surpasses 450 my, the variation is more likely to disrupt protein function.
SNPs&GO
SNPs&GO (https://snps.biofold.org/snps-and-go/snps-and-go.html) [31] is a single SVM-based prediction tool that takes protein sequences, profiles, and functional properties into account. It can detect if a mutation cause disease or not depending on the associated protein’s functional annotations. If RI (Reliability Index) result is > 0.5, the mutation is thought to be disease-related; otherwise, it is considered normal.
PROVEAN
PROVEAN ([32] evaluates if a protein's biological activity is affected by an AA alteration or indel. When the PROVEAN value is ≤ -2.5, the mutation is deleterious, and when it is > -2.5, it is expected to be neutral.
Meta-SNP
Meta-SNP (https://snps.biofold.org/meta-snp) [33] has been optimised to determine whether a specific point variant in polypeptide is disease related or belongs to the non-synonymous polymorphic (single-nucleotide variants) SNV category. The predictions of the four predictors are combined in Meta-SNP's output (PhD-SNP, SIFT, PANTHER, and SNAP). Meta-SNP provided SIFT (sorting intolerant from tolerant) results.
MUpro
MUpro (http://mupro.proteomics.ics.uci.edu) [34] is a set of computational algorithms that employ SVM and neural networks to assess the consequences of specific amino acid alterations on protein stability. If the score is 0 or lower, the variation has a negative impact on protein stability. However, when the score is higher than zero, the variation enhances the protein's stability.
INPS (Impact of Non-synonymous mutations on Protein Stability)
INPS (https://inpsmd.biocomp.unibo.it/inpsSuite/default/index2) [35] is a server that uses SVR (support vector regression) that predicts how a single-point variation will impact protein stability with reference to both sequence and structure of input protein.
DUET, SDM and mCSM
DUET (http://biosig.unimelb.edu.au/duet/stability) [36, 37] is a platform that uses a combination of computational methods to analyse missense mutations in protein structures. It combines the results of two complementing techniques (SDM and mCSM) to get a consensus prediction. As a result of the mutation, the analysis revealed the expected variance in folding free energy (ΔΔG in Kcal/mol). A negative number denotes a destabilising mutation, while a positive number denotes a stabilising mutation. The mCSM and SDM predictions for protein stability are shown separately.
Generation of in silico mutants and Molecular Dynamic Simulation
The mutation wizard of PyMOL (https://pymol.org/2) [38] was used to genarate an in silico mutants of identified nsSNPs in α-synuclein. MD Simulations of wild-type α-synuclein as well as its mutant structures were done to characterise functional and structural integrity, as well as to see if the mutants affect the protein's stability. We employed the GROMACS (Groningen Machine for Chemical Simulations) to perform 100 ns MD simulations using CHARMM27 force field settings [39,40,41,42]. The CHARMM27 protein force field is best for the intrinsically disordered proteins (IDPs) like α-synuclein as it increases the precision of conformational ensembles for IDPs. MD simulations were started with the structure of wild-type α-synuclein and its mutants G25S, V66E, and V77D structures created by PyMOL. The four-point TIP4P rigid water model has been used to solvate systems in a triclinic box of volume 1922.70 nm3 constructed through gmx editconf module. With the inclusion of counter ions (positive or negatively charged), the system was balanced, and energy minimization was attained using the steepest descent approach as shown in Table 1. The NPT and NVT thermostats were then used with a 1 bar Parrinello–Rahman isotropic pressure coupling and a 300 K Nose–Hoover temperature coupling for 3 ns. For both wild-type α-synuclein and its mutant structures, simulation runs of 100 ns were performed. Following the completion of MD simulations of wild-type α-synuclein and its mutant structures, we calculated a comprehensive study of structural variations in wild-type and mutant structures from the corrected trajectories (after the removal of PBC). The gmx rms, gmx rmsf, gmx gyrate, and gmx sasa tools were used to perform root-mean-square deviation (RMSD), root-mean-square fluctuations (RMSF), radius of gyration (Rg), and solvent accessible surface area (SASA) analysis. We also computed the variations caused in helix properties of structure of the wild type and mutants of α-synuclein by using gmx helix module of GROMACS.
In addition, we evaluated and analysed biologically relevant dominant motions by conducting essential dynamics based on principal component analysis of wild-type α-synuclein and its mutant structures. The MD simulation findings of wild-type α-synuclein and its mutant structures were evaluated using built-in functions of Gromacs, Bio3d package in R, [43] PyMOL and QtGrace.
Results
Primary sequence analysis
UniProt (https://www.uniprot.org) [17] database was used to extract the amino acid sequence of α-synuclein having Accession No. AAA16117.1. ProtParam (https:// web.expasy.org/protparam) [19] tool predicted different chemical and physical properties of the α-synuclein. It consists of 140 amino acid residues, predicted molecular mass of 14.5 kDa, predicted pI is 4.67, instability index of 25.47 means it is stable at room temperature, estimated half-life is greater than 20 h, aliphatic index is 69.64 that shows this protein is thermodynamically stable over a broad range of temperature and it also contains high amount of hydrophobic amino acid residues as predicted by its, grand average of hydropathicity (GRAVY) which is -0.403. The SMART (http://smart.embl-heidelberg.de) [20] web server predicted that the α-synuclein protein has no specific domains depending upon the sequence of amino acids. NetPhos 3.1 (https://services.healthtech.dtu.dk/service.php?NetPhos-3.1) [21, 22] and GPS 3.0 (http://gps.biocuckoo.org/online.php) [23] server anticipated phosphorylation of residues S9, T33, Y39, S42, T54, T59, T81, T92, Y125, S129, and Y133 in α-synuclein protein.
According to the subcellular location findings by TargetP 2.0 (https://services.healthtech.dtu.dk/service.php?TargetP-2.0) [24] and SignalP 5.0 (https://services.healthtech.dtu.dk/service.php?SignalP-5.0) [24] α-Synuclein is a secretory protein and has no signal peptide. TOPCONS (https://topcons.cbr.su.se) [25] showed that α-synuclein is not a homologous transmembrane protein.
STRING (https://string-db.org) [26] database identified both known and potential interacting partners of α-synuclein. They were HSPA8 (Heat shock protein member 8), TH, VAMP2 (Vesicle-associated membrane protein 2), PARK7 (Parkinson disease protein 7), TPPP (Tubulin polymerization-promoting protein), PARK2 (Parkin), HSPA4 (Heat shock 70 kDa protein 4), SLC6A3 (dopamine active transporter), SNCAIP (Synphilin-1 also known as synuclein, alpha interacting protein), and LRRK2 (Leucine-rich repeat kinase 2) as shown in Fig. 1.

Possible physical and functional interacting partners of α-synuclein identified by STRING database
Binding pocket analysis
CASTp was used to find probable binding sites and the binding site residues of α-synuclein. The best predicted pocket is composed of total 13 amino acid residues, whose solvent accessible surface area and pocket volume are 148.314 Å2 and 599.861 Å3, respectively, as shown in Table 2.
Deleterious nsSNPs prediction in coding region
Ensembl genome browser (https://asia.ensembl.org/index.html) [18] was used to collect the nsSNP data for α-synuclein. There were total 184 numbers of nsSNPs present in the sequence which were missense variants, out of which 10 were in the splice region. We employed various in silico tools to predict deleterious nsSNPs from 174 missense variants in the coding region.
Classification and evaluation of predicted deleterious nsSNPs by various in silico tools
In present study, I-Mutant 3.0(http://gpcr2.biocomp.unibo.it/cgi/predictors/I-Mutant3.0/I-Mutant3.0.cgi) [28] identified 88 nsSNPs with DDG value < 0 which may reduce protein stability. The DDG value is calculated from the unfolding Gibbs free energy value of the mutated protein minus the unfolding Gibbs free energy value of the wild type (Kcal/mol), PolyPhen-2(http://genetics.bwh.harvard.edu/pph2) [29] predicted 79 nsSNPs as probably damaging, PhD-SNP(https://snps.biofold.org/phd-snp/phd-snp.html) [28] with a score of > 0.5 predicted 25 nsSNPs to be disease causing, PANTHER (http://www.pantherdb.org/tools/csnpScoreForm.jsp) [30] identified 40 nsSNPs as probably damaging, SNPs&GO (https://snps.biofold.org/snps-and-go/snps-and-go.html) [31] predicted 87 nsSNPs as disease causing with a score of > 0.5 and PROVEAN (http://provean.jcvi.org/seq_submit.php) [32] anticipated 60 nsSNPs with a score of ≤ -2.5 as deleterious as shown in Fig. 2.

Identification and classification of deleterious nsSNPs in α-synuclein: (A) Flowchart describing the in silico procedure for evaluating deleterious nsSNPs and subsequent structural analysis (B) Bar diagram representing the number of nsSNPs predicted as deleterious by following in silico tools: I-Mutant 3.0 (88 nsSNPs), PolyPhen-2 (79 nsSNPs), PhD-SNP (25 nsSNPs), PANTHER (40 nsSNPs), SNPs&GO (87 nsSNPs), PROVEAN (60 nsSNPs)
Out of all these nsSNPs, three were found to be damaging by all in silico tools used. They are rs1433622151 (G25S), rs1261243630 (V66E), and rs745815563 (V77D) found as damaging by I-Mutant 3.0, PolyPhen-2, PhD-SNP, PANTHER, SNPs&GO, PROVEAN which are located in the predicted binding pocket. To determine the possible deleterious effects of these mutations, we used five additional in silico tools: MUpro, INPS, DUET, SDM, and mCSM. Tables 3 and 4(a) show the result. We also computed physicochemical properties of these 3 nsSNPs using ProtParam (https:// web.expasy.org/protparam) [19] as shown in Table 4(b).
Generation of in silico mutants
The mutation wizard of PyMOL was used in our study to genarate an in silico mutant structures of variations as shown in Table 5, that had been found deleterious by all in silico tools and are also located in predicted binding pocket. The variants are—rs1433622151 (G25S), rs1261243630 (V66E), and rs745815563 (V77D).
Molecular Dynamic (MD) Simulation
We performed MD simulations of 100 ns for wild-type α-synuclein and its mutant structures to further comprehend the structure and functional properties of the predicted disease-causing mutations. After the end of the MD simulations, the structural integrity and stability of wild-type and mutant structures was determined using the various analyses like root-mean-square deviation (RMSD), root-mean-square fluctuations (RMSF), radius of gyration (Rg), and solvent accessible surface areas (SASA). Effect of mutants on the helix properties of protein was also studied. Essential dynamics based on principal component analysis of wild-type and mutant α-synuclein structures were also used to study and understand biologically relevant motions.
The RMSD approach was used to evaluate the protein’s structural stability and its mutations on a consistent time frame [44]. RMSD assessment was carried out on all mutant structures as well as the wild type of α-synuclein. In both wild-type α-synuclein and its mutant structures, structural variations have been detected. Compared to the wild-type and the mutant structures, all three mutant structures showed significant deviation from wild-type α-synuclein as shown in Fig. 3a and Table 6. The average values of RMSD for wild type, G25S, V66E, and V77D were 3.28 nm, 2.60 nm, 2.90 nm, and 3.03 nm, respectively. The average of all the three mutations indicate that these variants alter the protein structure of α-synuclein. We used the RMSF to check if the amino acid alteration changed the dynamic characteristics of the residues. The flexible and non-flexible portions of wild-type and mutant α-synuclein structures can be identified using the RMSF parameter. In this analysis, we found fluctuations in protein structure with all the three mutations throughout the simulation up to 100 ns. When compared to wild-type structures, the V66E mutant structure displayed more fluctuations as shown in Fig. 3b and Table 6. The average RMSF values for wild type, G25S, V66E, and V77D were 0.66 nm, 1.15 nm, 1.51 nm, and 0.85 nm, respectively. The total RMSF data revealed that all three mutations show the increase in the fluctuations as compared to the wild-type structure of the protein. The radius of gyration (Rg) is explained as the mass-weight root-mean-square distance between a cluster of atoms and their shared centre of mass. As a result, it gives data on the protein’s overall dimension. In this study, we found that the Rg value for G25S was higher as compared to V66E and V77D as shown in Fig. 3c and Table 6. The average Rg values for wild type, G25S, V66E, and V77D were 2.61 nm, 4.06 nm, 2.99 nm, and 2.59 nm, respectively. The Rg findings suggest that though G25S shows higher values, all three mutations influence folding pattern of wild-type structure. We applied SASA to investigate wild-type and mutant α-synuclein structures to determine the solvent accessibility of α-synuclein and the implications of mutations on the solvent effect of α-synuclein as shown in Fig. 3d and Table 6. The average SASA values for wild type, G25S, V66E, and V77D were 115.24 nm2, 128.14nm2, 128.91nm2, and 118.7nm2, respectively. The SASA values of mutant structures are higher than native form revealed that due to the influence of mutations structural changes occurred.

Molecular dynamics simulation results of the wild-type α-synuclein and 3 mutant (V77D, V66E, and G25S) structures of α-synuclein. (a) Root-mean-square deviation, (b) Root-mean-square fluctuation, (c) Radius of gyration and (d) Solvent accessible surface area. Wild type is represented in black, mutant G25S in red, mutant V66E in green, and mutant V77D in blue
The essential dynamics (ED) approach was utilised to capture the fundamental, biologically relevant motions from the global trajectories of wild-type and mutant α-synuclein structures for the principal component analysis. In ED, two variables, PC1 (Principal Component 1) and PC2 (Principal Component 2), measure the aggregate fluctuations of the most variable areas of α-synuclein. The eigenvalue ranks and two-dimensional projections of the significant variations are depicted in Fig. 4. The initial conformations of wild type and mutants are reported by blue dots, whereas the intermediate and final conformations are represented by the white and red dots, respectively. The continuous colour scale from blue to white to red dots indicates that there were periodic jumps between these conformers throughout the trajectory. The projections implied that, in comparison with the wild-type α-synuclein, the mutants show distinct conformational changes in the essential dynamics of α-synuclein. The initial conformations of wild-type α-synuclein protein are clustered in left and right top side of essential subspace while the final conformations are tightly clustered in bottom right of essential subspace. The conformational subspace occupied by wild-type α-synuclein along PC1 is 67.14% and 7.66% for PC2. Further, the conformational subspace occupied mutant G25S along PC1 is 37.36% and 23.58% for PC2. Similarly, the conformational subspace occupied by mutant V66E along PC1 is 56.66% and 20.94% for PC2. Lastly, the conformational subspace occupied by mutant V77D along PC1 is 54.81% and 12.74% for PC2. By PCA graphs, we found that all mutants show higher flexibility than wild type as shown in Fig. 4. The total PCA analysis revealed that all mutants affect the stability of α-synuclein protein, which line with the observations of all the initial analysis. Moreover, from the PCA analysis, it has been observed that the trace of covariance matrix values is altered in comparison with native form which again indicated that missense mutations are significantly alter the conformational behaviour of alpha synuclein protein.

Principal component (essential dynamics) analysis of the wild-type α-synuclein and mutant G25S, V66E, and V77D structures of α-synuclein. (a) wild-type α-synuclein, (b) G25S mutant, (c) V66E mutant and (d) V77D mutant
We also calculated variations caused in helix properties of structure of the wild type and mutants of α-synuclein by using gmx helix module of GROMACS. In this analysis, various helical properties like helix radius, twist, rise per residue, total helix length, RMS deviation from ideal helix, average phi-psi angles, helicity per residue, and average Calpha—Calpha dihedral angle were computed to understand the effect of the mutation on the wild-type α-synuclein protein. The various values obtained by these analysis are shown in Table 7. All helix properties showed alterations in all mutants as compared to wild-type α-synuclein.The graphs showing the variations in different helix properties are given in Additional file 1.
Overall molecular dynamics simulation results show that the mutations affect the structural stability and dynamic behaviour of the α-synuclein protein. Therefore, using a molecular dynamics simulation we compared the structures of wild type and all three mutants of α-synuclein protein in this study. Throughout the 100 ns simulation, structural alterations were seen in all three mutants of the alpha-synuclein protein as compared to the wild type. These nsSNPs of the α-synuclein protein may destabilise its structure, which may have an impact on its function, according to this molecular dynamics study. If the activity of α-synuclein protein is disrupted, its cellular functioning may be compromised, leading to serious human malfunctions such neurodegenerative illnesses.
4. Discussion
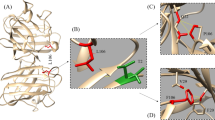
This present study to pinpoint the most harmful genetic variations in α-Synuclein and understand how they affect the protein's stability. By employing various specialised computer techniques related to diseases, we could separate harmful genetic variations from those that are harmless or neutral. We conducted an exhaustive search using a SNP database adhering to well-defined criteria to ensure accuracy and reliability and identified 174 missense nsSNPs. Various computational tools were then employed to assess their harmfulness. Through analysis using seven different in silico tools (I-Mutant 3.0, PolyPhen-2, PhD-SNP, PANTHER, SNPs&GO, PROVEAN, SIFT), three nsSNPs, specifically rs1433622151 (G25S), rs1261243630 (V66E), and rs745815563 (V77D), were consistently identified as disease causing or deleterious variants. Three-dimensional models of the nsSNP variations G25S, V66E, and V77D were generated to evaluate the structural stability of the mutant proteins in comparison with the wild-type protein. The Ramachandran plot, utilised for assessing the structural precision of protein models, confirmed the validity of our predicted variant models. The structural stability and dynamic behaviour of all three mutants, as well as the wild-type α-synuclein, were examined through 100 ns of molecular dynamics simulations. This allowed for a comprehensive assessment of how variations affected the protein's structural integrity, particularly highlighting the dynamic differences between the mutants and the wild-type α-synuclein. Subsequent analyses following the molecular dynamics simulations concluded that all three mutations substantially destabilised the structural integrity of the wild-type protein. Specifically, the G25S, V66E, and V77D mutants were found to disrupt the stability of the wild-type protein, potentially interfering with its function. These mutations were observed to exert a notable influence on the structural stability and dynamic behaviour of the α-synuclein protein. The lipid binding domain of α-synuclein spans from residue number 1 to 95, characterised by its hydrophobic nature and its involvement in protein aggregation [4, 5]. Notably, all three mutations—G25S, V66E, and V77D—are situated within this lipid binding domain of α-synuclein, as illustrated in Fig. 5. Single-nucleotide polymorphisms (SNPs) occurring in crucial domains of proteins can cause disruptions in their structure, leading to functional impairment. In the case of α-synuclein, all three predicted nsSNPs are located within the hairpin structure of the protein. This positioning has the potential to disrupt intermolecular interactions necessary for tetramer formation. Consequently, an elevated presence of monomeric α-synuclein may occur, contributing to its aggregation, which is implicated in neurodegeneration [45]. The substitution of glycine with serine at the 25th position induces structural alterations in α-synuclein. Glycine typically does not participate in post-translational modifications (PTMs) on its side chain, whereas serine is commonly engaged in numerous PTMs, including O-glycosylation, O-acetylation, O-octanoylation, O-palmitoylation, N-acetylation, N-decanoylation, and notably, phosphorylation. These PTMs can variably modify the structural interactions and functional activities of α-synuclein [46]. Valine is an amino acid with hydrophobic properties, while glutamic acid and aspartic acid are hydrophilic in nature. Alterations in post-translational modifications (PTMs) are often indicative of disease states. The transition from a hydrophobic to a hydrophilic amino acid can significantly impact the lipid interactions of α-synuclein. Valine residues at positions 66 and 77 are situated within the NAC region of α-synuclein, which possesses the ability to interact with lipids due to the hydrophobic nature of valine. The substitution of valine with hydrophilic amino acids like glutamic acid and aspartic acid may result in the loss of interactions with lipids, consequently promoting the formation of beta aggregates and disruption of membranes [4]. Furthermore, changes in α-synuclein's membrane-binding capabilities may also impact its physiological function in processes such as vesicle trafficking and neurotransmitter release, which rely on the protein's interaction with lipid membranes. This study examines the potential roles of the G25S, V66E, and V77D variants of α-synuclein in neurodegenerative disorders, aiming to uncover their contributions to disease progression and pave the way for further investigation in this field. In essence, this study represents a significant step forward in our understanding of the structural and functional implications of genetic variations in α-synuclein, particularly in the context of neurodegenerative disorders. By elucidating the intricate molecular mechanisms underlying these variations, the study not only sheds light on the pathogenesis of these devastating diseases but also lays the groundwork for future investigations into potential therapeutic interventions targeting α-synuclein dysfunction.

Location of predicted nsSNPs on the sequence of α-Synuclein
Conclusion
SNPs represent one of the most prevalent risk factors across a range of complex illnesses, affecting the structure and function of resulting proteins within the protein-coding region. Various methods have been utilised to explore the deleterious effects associated with nsSNPs in numerous disorders. Presently, computational analysis has generated a roadmap for identifying typical disease-specific nsSNPs at the molecular level. In this current study, we have identified three highly critical deleterious variants, G25S, V66E, and V77D, within the amino acid sequence of α-synuclein. To examine the structural and functional consequences of these predicted deleterious nsSNPs, a 100 ns molecular dynamics simulation was performed. Our results indicate that the G25S nsSNP could promote serine-dependent phosphorylation of α-synuclein, impacting its structural stability. Similarly, the V66E and V77D nsSNPs alter the hydrophobic to hydrophilic amino acid composition, significantly affecting α-synuclein's interaction with lipids. These alterations in lipid binding may disrupt tetramer formation, leading to α-synuclein aggregation, ultimately contributing to neurodegeneration. These structural modifications may play a crucial role in neurodegenerative diseases. The prioritised nsSNPs identified in the present study could serve as valuable targets for further experimental research, particularly, by site directed mutagenesis (SDM), enabling the assessment of α-synuclein protein mutations in relation to neurodegenerative disorders and facilitating the development of therapeutics against these mutant forms of α-synuclein.
Availability of data and materials
All data generated or analysed during this study are included in this manuscript.
Abbreviations
- α-Synuclein:
-
Alpha synuclein
- nsSNPs:
-
Non-synonymous single-nucleotide polymorphisms
- PD:
-
Parkinson’s disease
- ΔG :
-
Gibbs free energy
- GROMACS:
-
Groningen Machine for Chemical Simulations
- RMSF:
-
Root-mean-square fluctuations
- RMSD:
-
Root-mean-square deviation
- Rg:
-
Radius of gyration
- SASA:
-
Solvent accessible surface area
- PCA:
-
Principal component analysis
- MD:
-
Molecular dynamics
- ED:
-
Essential dynamic
References
Fung KM, Rorke LB, Giasson B, Lee VM, Trojanowski JQ (2003) Expression of alpha-, beta-, and gamma-synuclein in glial tumors and medulloblastomas. Acta Neuropathol 106(2):167–175. https://doi.org/10.1007/s00401-003-0718-x
George JM (2002) The synucleins. Genome Biol 3(1):52. https://doi.org/10.1186/gb-2001-3-1-reviews3002
van Rooijen BD, van Leijenhorst-Groener KA, Claessens MM, Subramaniam V (2009) Tryptophan fluorescence reveals structural features of alpha-synuclein oligomers. J Mol Biol 394(5):826–833. https://doi.org/10.1016/j.jmb.2009.10.021
Emamzadeh FN (2016) Alpha-synuclein structure, functions, and interactions. J Res Med Sci 21:29. https://doi.org/10.4103/1735-1995.181989
Uchihara T, Giasson BI (2016) Propagation of alpha-synuclein pathology: hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol 131(1):49–73. https://doi.org/10.1007/s00401-015-1485-1
Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC (2010) Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science (New York, N. Y.) 329(5999):1663–1667. https://doi.org/10.1126/science.1195227
Chandra S, Chen X, Rizo J, Jahn R, Südhof TC (2003) A broken alpha -helix in folded alpha -Synuclein. J Biol Chem 278(17):15313–15318. https://doi.org/10.1074/jbc.M213128200
Sun J, Wang L, Bao H, Premi S, Das U, Chapman ER, Roy S (2019) Functional cooperation of α-Synuclein and VAMP2 in synaptic vesicle recycling. Proc Natl Acad Sci USA 116(23):11113–11115. https://doi.org/10.1073/pnas.1903049116
Atias M, Tevet Y, Sun J, Stavsky A, Tal S, Kahn J, Roy S, Gitler D (2019) Synapsins regulate α-Synuclein functions. Proc Natl Acad Sci USA 116(23):11116–11118. https://doi.org/10.1073/pnas.1903054116
Filippini A, Gennarelli M, Russo I (2019) α-Synuclein and glia in Parkinson’s disease: a beneficial or a detrimental duet for the endo-lysosomal system? Cell Mol Neurobiol 39(2):161–168. https://doi.org/10.1007/s10571-019-00649-9
Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VM, Trojanowski JQ, Iwatsubo T (1998) Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am J Pathol 152(4):879–884
Bousset L, Pieri L, Ruiz-Arlandis G, Gath J, Jensen PH, Habenstein B, Madiona K, Olieric V, Böckmann A, Meier BH, Melki R (2013) Structural and functional characterization of two alpha-synuclein strains. Nat Commun 4:2575. https://doi.org/10.1038/ncomms3575
Chu Y, Dodiya H, Aebischer P, Olanow CW, Kordower JH (2009) Alterations in lysosomal and proteasomal markers in Parkinson’s disease: relationship to alpha-synuclein inclusions. Neurobiol Dis 35(3):385–398. https://doi.org/10.1016/j.nbd.2009.05.023
Seidel K, Schöls L, Nuber S, Petrasch-Parwez E, Gierga K, Wszolek Z, Dickson D, Gai WP, Bornemann A, Riess O, Rami A, Den Dunnen WF, Deller T, Rüb U, Krüger R (2010) First appraisal of brain pathology owing to A30P mutant alpha-synuclein. Ann Neurol 67(5):684–689. https://doi.org/10.1002/ana.21966
George PDC, Rajith B (2012) Computational refinement of functional single nucleotide polymorphisms associated with ATM gene. PloS one 7(4):e34573. https://doi.org/10.1371/journal.pone.0034573
Singh RK, Mahalingam K (2017) In silico approach to identify non-synonymous SNPs in human obesity related gene, MC3R (melanocortin-3-receptor). Comput Biol Chem 67:122–130. https://doi.org/10.1016/j.compbiolchem.2016.12.009
UniProt Consortium (2021) UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res 49(D1):D480–D489. https://doi.org/10.1093/nar/gkaa1100
Howe KL, Achuthan P, Allen J, Allen J, Alvarez-Jarreta J, Amode MR, Armean IM, Azov AG, Bennett R, Bhai J, Billis K, Boddu S, Charkhchi M, Cummins C, Da Rin FL, Davidson C, Dodiya K, El Houdaigui B, Fatima R, Gall A, Flicek P (2021) Ensembl 2021. Nucleic Acids Res 49(D1):D884–D891. https://doi.org/10.1093/nar/gkaa942
Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD, Hochstrasser DF (1999) Protein identification and analysis tools in the ExPASy server. Methods Mol Biol 112:531–552. https://doi.org/10.1385/1-59259-584-7:531
Letunic I, Khedkar S, Bork P (2021) SMART: recent updates, new developments and status in 2020. Nucleic Acids Res 49(D1):D458–D460. https://doi.org/10.1093/nar/gkaa937
Blom N, Gammeltoft S, Brunak S (1999) Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol 294(5):1351–1362. https://doi.org/10.1006/jmbi.1999.3310
Blom N, Sicheritz-Pontén T, Gupta R, Gammeltoft S, Brunak S (2004) Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 4(6):1633–1649. https://doi.org/10.1002/pmic.200300771
Wang C, Xu H, Lin S, Deng W, Zhou J, Zhang Y, Shi Y, Peng D, Xue Y (2020) GPS 5.0: an Update on the Prediction of Kinase-specific Phosphorylation Sites in Proteins. Genom Proteom Bioinfor 18(1):72–80. https://doi.org/10.1016/j.gpb.2020.01.001
Almagro AJJ, Tsirigos KD, Sønderby CK, Petersen TN, Winther O, Brunak S, von Heijne G, Nielsen H (2019) SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat Biotechnol 37(4):420–423. https://doi.org/10.1038/s41587-019-0036-z
Tsirigos KD, Peters C, Shu N, Käll L, Elofsson A (2015) The TOPCONS web server for consensus prediction of membrane protein topology and signal peptides. Nucleic Acids Res 43(W1):W401–W407. https://doi.org/10.1093/nar/gkv485
Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, Doncheva NT, Legeay M, Fang T, Bork P, Jensen LJ, von Mering C (2021) The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res 49(D1):D605–D612. https://doi.org/10.1093/nar/gkaa1074
Tian W, Chen C, Lei X, Zhao J, Liang J (2018) CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Res 46(1):363–367. https://doi.org/10.1093/nar/gky473
Capriotti E, Calabrese R, Casadio R (2006) Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics (Oxford, England) 22(22):2729–2734. https://doi.org/10.1093/bioinformatics/btl423
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR (2010) A method and server for predicting damaging missense mutations. Nat Methods 7(4):248–249. https://doi.org/10.1038/nmeth0410-248
Mi H, Ebert D, Muruganujan A, Mills C, Albou LP, Mushayamaha T, Thomas PD (2021) PANTHER version 16: a revised family classification, tree-based classification tool, enhancer regions and extensive API. Nucleic Acids Res 49(D1):D394–D403. https://doi.org/10.1093/nar/gkaa1106
Capriotti E, Altman RB (2011) Improving the prediction of disease-related variants using protein three-dimensional structure. BMC Bioinf 12(4):3. https://doi.org/10.1186/1471-2105-12-S4-S3
Choi Y, Sims GE, Murphy S, Miller JR, Chan AP (2012) Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 7(10):e46688. https://doi.org/10.1371/journal.pone.0046688
Capriotti E, Altman RB, Bromberg Y (2013) Collective judgment predicts disease-associated single nucleotide variants. BMC Genom. https://doi.org/10.1186/1471-2164-14-S3-S2
Cheng J, Randall A, Baldi P (2006) Prediction of protein stability changes for single-site mutations using support vector machines. Proteins 62(4):1125–1132. https://doi.org/10.1002/prot.20810
Savojardo C, Fariselli P, Martelli PL, Casadio R (2016) INPS-MD: a web server to predict stability of protein variants from sequence and structure. Bioinformatics (Oxford, England) 32(16):2542–2544. https://doi.org/10.1093/bioinformatics/btw192
Pires DE, Ascher DB, Blundell TL (2014) mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics (Oxford, England) 30(3):335–342. https://doi.org/10.1093/bioinformatics/btt691
Worth CL, Preissner R, Blundell TL (2011) SDM-a server for predicting effects of mutations on protein stability and malfunction. Nucleic Acids Res 39:W215–W222. https://doi.org/10.1093/nar/gkr363
Schrödinger LLC (2015) The PyMol molecular graphics system, Versión 1.8. Thomas Holder 1(1):1–5
Páll S, Abraham MJ, Kutzner C, Hess B, Lindahl E (2015) Tackling exascale software challenges in molecular dynamics simulations with GROMACS. In Markidis S, Laure E (eds) Solving Software Challenges for Exascale. EASC 2014. Lecture Notes in Computer Science, vol 8759. Springer, Cham. https://doi.org/10.1007/978-3-319-15976-8_1
Van der Spoel D, Hess B (2011) GROMACS-the road ahead. Wires Comput Mol Sci 1(5):710–715. https://doi.org/10.1002/wcms.50
Bjelkmar P, Larsson P, Cuendet MA, Hess B, Lindahl E (2010) Implementation of the CHARMM force field in GROMACS: analysis of protein stability effects from correction maps, virtual interaction sites, and water models. J Chem Theory Comput 6(2):459–466. https://doi.org/10.1021/ct900549r
Huang J, MacKerell AD Jr (2018) Force field development and simulations of intrinsically disordered proteins. Curr Opin Struct Biol 48:40–48. https://doi.org/10.1016/j.sbi.2017.10.008
Grant BJ, Rodrigues APDC, Elsawy KM, Mccammon AJ, Caves LSD (2006) Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics 22:2695–2696
Kuzmanic A, Zagrovic B (2010) Determination of ensemble-average pairwise root mean-square deviation from experimental B-factors. Biophys J 98(5):861–871. https://doi.org/10.1016/j.bpj.2009.11.011
Kara E, Lewis PA, Ling H, Proukakis C, Houlden H, Hardy J (2013) α-Synuclein mutations cluster around a putative protein loop. Neurosci Lett 546:67–70. https://doi.org/10.1016/j.neulet.2013.04.058
Uversky VN (2015) The intrinsic disorder alphabet. III. Dual personality of serine. Intrinsically DisordProteins 3(1):e1027032. https://doi.org/10.1080/21690707.2015.1027032
Lyra P, Machado V, Rota S, Chaudhuri KR, Botelho J, Mendes JJ (2023) Revisiting alpha-Synuclein pathways to inflammation. Int J Mol Sci 24(8):7137. https://doi.org/10.3390/ijms24087137
Srinivasan E, Chandrasekhar G, Chandrasekar P, Anbarasu K, Vickram AS, Karunakaran R, Rajasekaran R, Srikumar PS (2021) Alpha-Synuclein aggregation in Parkinson’s disease. Front Med 8:52
Yonova-Doing E, Atadzhanov M, Quadri M, Kelly P, Shawa N, Musonda ST, Simons EJ, Breedveld GJ, Oostra BA, Bonifati V (2012) Analysis of LRRK2, SNCA, Parkin, PINK1, and DJ-1 in Zambian patients with Parkinson’s disease. Parkinsonism Relat Disord 18(5):567–571. https://doi.org/10.1016/j.parkreldis.2012.02.018
Acknowledgements
The authors express their gratitude to Sharda University, Greater Noida, India.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
MJ and JM conceived the concept of the study. AS, PM, and MJ wrote the manuscript. AS, PM, and MJ prepared the figures and tables. AS, PM, and MJ collected the literature from various resources. MJ, JM, and AKS corrected the manuscript. All authors approved the contents of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. The results of helicity analysis of wild-type and mutant forms of Alpha Synuclein.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sharma, A., Mahur, P., Singh, A. et al. Insights into the structural and functional analysis of impact of the missense mutations on α-synuclein: an in silico study. Egypt J Med Hum Genet 25, 60 (2024). https://doi.org/10.1186/s43042-024-00530-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-024-00530-5