
Abstract
Objective
Biotinidase deficiency (BTD) is characterized by a wide range of genetic variants. However, the correlation between these variants and the biochemical phenotypes of BTD is not well-established due to the diversity of the BTD gene, the variable nature of biotinidase, and difficulties in measuring enzyme activity. This study aims to identify BTD gene variants in newborns screened for biotinidase deficiency in Southeastern Anatolia and to examine the correlation between these variants and biochemical phenotypes.
Materials and methods
BTD variant analysis and biotinidase enzyme (BT) activity measurements were performed on 711 newborns. Enzyme activity was measured using the colorimetric method. Biochemical phenotyping was categorized into three groups based on mean residual enzyme activity: profound (≤ 10%), partial (10.1–30%), and normal (> 30.1%). The pathogenicity of BTD gene variants was determined using BTD databases.
Results
The biochemical phenotypes were distributed as follows: a) profound: n = 22 (3%), b) partial: n = 95 (13.3%), and c) normal: n = 594 (83.7%). The mean enzyme activities (%) for these groups were 8.79 ± 1.87, 22.67 ± 4.55, and 97.98 ± 17.45, respectively. The most common alleles and their frequencies were p.D444H (n = 526) (37%), p.R157H (n = 172) (12.1%), and p.C33Ffster*36 (n = 73) (9%). The pathogenicity of the variants was as follows: pathogenic: 481 (33.8%), likely pathogenic: 4 (0.2%), and variant of uncertain significance (VUS): 538 (37.8%).
Conclusion
In this large cohort in Southeastern Anatolia, the most common alleles were p.D444H, p.R157H, and p.C33Ffster*36 in BTD variants. The results indicate a low concordance between the biochemical phenotype and genotype in newborns with BTD. This study highlights the inadequacy of predicting the biochemical phenotype based solely on variant pathogenicity in biotinidase deficiency during the neonatal period.
Similar content being viewed by others


Introduction
Biotinidase deficiency (BTD) is one of the most common autosomal recessive diseases globally, with an incidence of 1:40,000–1:60,000 per live birth [1]. However, in Turkey, where consanguineous marriages are widespread, the frequency reaches up to 1:7116. Over 200 variants have been identified in the BTD gene [2]. BTD can cause neurodevelopmental delay, seizures, visual loss, sensorineural hearing loss, and rash in children of any age, from birth to adulthood. Biotinidase activity is frequently measured using the calorimetric technique. Based on residual enzyme activity, three biochemical phenotypes are categorized as follows: profound (≤ 10%), partial (10.1–30%), and normal (> 30.1%). However, the biochemical phenotypes can exhibit variability. Biotinidase activity does not act along a continuous spectrum [3]. Consequently, phenotypes may not always match the expected genotype when phenotypes are identified using predetermined cut-off values. Biotinidase activity has biphasic kinetics, which can lead to clinical heterogeneity [4]. In addition, enzyme maturity develops with age [5]. Screening tests for biotinidase are typically carried out three to five days after birth, at the lowest point of the enzyme’s activity. Therefore, biochemical phenotypic groups identified in the early neonatal period can change. These factors make it challenging to evaluate neonatal screening results, and newborns are frequently overdiagnosed with BTD. This complexity can make it challenging for each patient to choose a suitable therapy strategy and raise questions about cost-effectiveness [6].
The BTD gene, located on chromosome 3 and consisting of 4 exons and 543 amino acids, encodes biotinidase. BTD is caused by pathogenic variants in the BTD gene, resulting in a variety of clinical phenotypes [7]. Over 200 pathogenic variants have been identified, with the number of newly reported variants increasing. The frequency and diversity of disease-causing variants vary by country and region. The most common variants are generally: p.D444H (c.1330G > C), p.R157H (c.470G > A), p.C33Ffster*36 (c.198_104delGCGGCTGinsTCC), p.(T532M) (c.1595C > T), and p.Q456H ( c.1368A > C) (7). However, the correlation between genotype and phenotype is not well-established. The diversity of variant allele combinations also affects clinical and biochemical phenotypes. Despite some severe pathogenic variants exhibiting predictable clinical phenotypes, the genotype–phenotype relationship in BTD is not well-established.
This large cohort study aimed to investigate the relationship between BTD variants and biochemical phenotypes.
Materials and methods
This study was conducted at the Department of Pediatric Metabolism, Gaziantep University, as a single-center, observational, cross-sectional study. Approval for the study was obtained from the Clinical Research Ethics Committee of Gaziantep University (Decision no/date: 2019/194–19.6.2019). Written consent was obtained from all parents. Blood samples were collected on filtered paper three to five days after birth, following the national screening protocol. Patients who tested positive for biotinidase screening sought confirmation at our center within the first month of birth. Confirmation tests were conducted using quantitative measurements and the colorimetric method. Molecular analysis was performed to identify pathogenic variants of the BTD gene for each subject.
Subjects
The study included 711 nonrelative newborns referred to the Neonatal Biotinidase Screening Program. The majority of initial confirmatory tests were conducted within the first month of life. Oral biotin treatments of 10 mg/day (Profound) and 5 mg/day (Partial) were administered based on enzyme activity levels.
Enzyme activity measurement and determination of biochemical phenotype groups
Quantitative enzyme activity was measured using the colorimetric method. The normal reference range was 3.5–13.8 nmol/min/mL. Biochemical phenotyping was divided into three groups based on mean enzyme activity (7.5 nmol/min/mL): profound (≤ 10% of mean activity), partial (10.1–30%), and normal (> 30.1%).
Molecular analysis
Blood sample was sequenced using Sanger sequence method (AB 3130 XL) after amplification with PCR (Maxwell RSC Whole Blood DNA kit Promega, USA). DNA density was adjusted to 10–30 ng/ml using a spectrophotometer (Biotech USA). Sanger sequencing was conducted to analyze all exons, introns, and junctions of the BTD gene using the AB3130XL device. The genomic reference for the BTD gene in Ensembl was NM_000060(NM_001370658.1) (Transcript ID: ENS00000303498.5; Reference sequence F: ATGGCGCAT R:TACCGCGTA).
Pathogenic variants were classified according to their pathogenicity using the PubMed and BTD databases (ClinVar, Varsome, ARUP, LOVD, Ensemble, MutationTaster, ACMG). The clinical significance and variant pathogenicity of the BTD gene were assessed using BTD databases [8,9,10,11,12,13]. Variant pathogenicity was classified according to the ACMG criteria [14]. Novel pathogenic variants were defined as those not present in the mentioned databases.
Genotypes were classified as homozygous (HMZ), heterozygous (HTZ), and compound heterozygous (C/H) based on heterozygosity.
Statistics
Statistical analyses were performed using the Statistical Package for Social Sciences version 22.0 (IBM Inc., Chicago, IL, USA). The Shapiro–Wilk test was used to confirm the normal distribution of continuous variables. The chi-square test was used to determine the significance between the expected and observed enzyme activity groups.
Results
Among participants (n = 711), 325 (45.8%) were female. The distribution of biochemical phenotype groups based on biotinidase activity was as follows: profound 22 (3%), partial 95(13%), and normal 594 (84%), (Table 1). The mean age at the time of the test was 18.53 ± 8.54 days. Table 1 shows the mean levels of biotinidase activity for each phenotype group.
Of the total alleles (n = 1422), 386, 612 and 314 were from patients based on zygosity of 193 HMZ (27.1%), 306 HTZ (43.1%) and 157 C/H (22.1%), respectively. No pathogenic variant was detected in 55 (7.7%) patients (Table 1). Of these, 53 were in normal biochemical phenotype, while the remaining two were in the partial (n = 1) and profound (n = 1) groups.
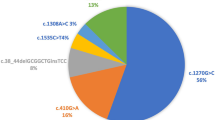
The most common alleles among the total alleles (n = 1422) were p.D444H (c.1330G > C) (n = 526,37%). The next two alleles with the highest frequencies were p.R157H (c.470G > A) (n = 172, 12.1%) and p.C33Ffster*36 (c.198_104delGCGGCTGinsTCC (n = 84, 6%) (Table 2). The other most frequent alleles are shown in Table 2. There were 11 cases with the p.D444H HMZ genotype that presented with Profound (n = 3) and Partial (n = 8) BTD. Less frequent alleles are given in Table 2. Six new previously unreported mutations were detected in the BTD databases (Table 2).
According to BTD databases, the pathogenicity classification of variants was as follows: pathogenic: 481 (33.8%), variant of uncertain significance (VUS): 538 (37.8%), 4 likely pathogenic (0.3%) (Table 1). The distribution of newborns with pathogenic variants in both alleles into biochemical phenotypes was as follows: profound: 15 (68%), partial: 29 (30.5%), and normal: 43 (7%). The distribution of pathogenic variants (n = 481) into profound, partial, and normal biochemical groups was as follows: pathogenic variant 43 (7%), 105 (22%), and 342 (71%). VUS (n = 538): 6 (1.1%), 70 (13%), and 462 (85.9%).
Discussion
This large cohort study examines the relationship between BTD variants and biochemical phenotypes during the neonatal period. Most of the reports related to BTD are cross-sectional studies and involve the neonatal or early infant period [15,16,17].
Previous studies on early infancy have reported a higher frequency of profound and partial BTD [15, 17, 18]. Biotinidase enzyme activity levels are reported to be the lowest in the first days of life, but increases within the following days [15]. Therefore, low levels of enzyme activity can be expected in early neonatal confirmation tests with screening performed in the first postnatal period. Biotinidase activity maturation later in life should be taken into account when assessing biochemical phenotyping based on biotinidase levels in newborns with BTD. However, there is not enough research on the course of low activity levels later in life. Considering this pattern of enzyme activity that has settled to normal levels at a later age, the relatively high frequency of BTD may reflect the effort to adapt to the transition from the intrauterine period to the extrauterine period rather than a disease. This variability in biotinidase activity is influenced by factors arising from various clinical conditions and the natural course of the disease. These include premature birth, environmental temperature, transfer conditions of the samples, hyperbilirubinemia, and perinatal asphyxia [11,12,13]. These findings indicate that a single measurement in the neonatal period is not sufficient to determine the current enzyme activity level, and follow-up with sequential tests is needed to assign biochemical phenotypes in BTD.
The most common variants found in this study were p.D444H, p.R157H, and p.C33Ffs. Although p.D444H is considered a mild variant, it was found to be associated with profound activity in three HMZ cases (p.D444H-p.D444H), partial enzyme activity in eight cases with HMZ (p.D444H-p.D444H), and partial enzyme activity in three HTZ cases (p.D444H-No). The p.D444H variant has, against expectations, also been documented in previous studies, where it has been correlated to more severe biochemical phenotypes [17,18,19,20,21]. This could be the result of double pathogenic variants, which are combinations of severe variants that are undetectable, or the additional contribution of epigenetic factors [22]. Given that frequent consanguineous marriages are common in our region, complex pathogenic variants appear to be more frequent [23, 24]. p.R157H was one of the most common severe variants. However, biotinidase activity was normal level in two patients with the HMZ p.R157H genotype. It was found that the p.C33Ffs variant was seen more frequently in this study, compared to other reports [17, 18]. It has been reported that the p.C33Ffs variant is one of the most common reasons for profound BTD in the USA [25]. Contrary to expectations, the normal enzyme activity group was detected in 12 p.C33Ffs heterozygous genotype (p.C33Ffs-No) cases. Unlike the expected phenotype, partial enzyme activities were observed in severe mutant alleles, such as p.Q456H and p.T532M.
The genotype–biochemical phenotype compatibility in this study was lower than previously reported data [7, 18]. This may be due to increased enzyme maturation and activity with age.
Many asymptomatic cases with severe pathogenic variants have been reported in the literature, and some viewpoints have been proposed for genotype–phenotype incompatibility [26]. Genotype–phenotype incompatibility suggests that there is sufficient residual enzyme activity in the presence of a substrate alongside epigenetic factors. Another view suggests that biotinidase is caused by individual differences in Km kinetics. Adequate dietary biotin intake may also help to prevent the appearance of clinical findings [27]. These asymptomatic cases may present with clinical findings such as acute hearing loss as a result of exposure to metabolic stress at later ages.
Preanalytical factors, on the other hand, such as blood sample transportation or storage conditions until analysis, may also affect the measurement of enzyme activity. These variables are limitations of these studies involving genotype–phenotype correlation in BTD. This study supports the hypothesis that variants identified as pathogenic in BTD databases alone cannot predict clinical phenotypes. Of the cases with normal enzyme activity, 7.2% had pathogenic variants in both alleles. These cases require long-term follow-up. Likewise, pathogenic variants in both alleles were present in 30.5% of the partial group. Similar cases have been reported in the literature [28]. Asymptomatic cases are described, despite the fact that they have pathogenic variants. This large cohort will contribute to the implications of these cases. Another important contribution of the study is that it reports six new pathogenic variants that were not previously identified in BTD databases. One of the study’s major strengths is that it is one of the largest cohorts in the literature dealing with the genotype–phenotype relationship in BTD patients. Another significant contribution is the identification of six new pathogenic variants.
Fifty-three of the 55 cases without any pathogenic variant had normal enzyme activity. These cases were patients who applied because of screening positivity. It was thought that the false positivity in these cases was due to the lower enzyme activity in the neonatal period. Confirmatory tests from these newborns were performed at the end of the first month. It was interpreted as the presence of an undetected variant in the other two newborns, one of which was partial and the other profound.
Conclusion
The most common alleles found in this large cohort in Southeastern Anatolia were p.D444H (c.1330G > C), p.R157H (c.470G > A), and p.C33Ffster*36. The results of this study show that there is a low concordance between genotype and biochemical phenotype in newborns with BTD in this region. This study confirms that it is not sufficient to establish a predictable correlation between BTD variant pathogenicity and biochemical phenotype in newborns with biotinidase deficiency. Additionally, this study emphasizes the importance of considering clinical features and addressing analytical difficulties in enzyme activity measurements and molecular analyses to explain the mismatch between genotype and biochemical phenotype.
Availability of data and materials
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Wolf B (1991) Worldwide survey of neonatal screening for biotinidase deficiency. J Inherit Metab Dis 14:923–927. https://doi.org/10.1007/BF01800475
Procter M, Wolf B, Crockett D, Mao R (2013) The biotinidase gene variants registry: a paradigm public database. G3 Bethesda. 3:727–731. https://doi.org/10.1534/g3.113.005835
Cowan M, Blitzer MG, Wolf B (2010) Technical standards and guidelines for the diagnosis of biotinidase deficiency. Genet Med 12:464–470. https://doi.org/10.1097/GIM.0b013e3181e4cc0f
Ramaekers T, Suormala M, Brab M, Duran R, Heimann G, Baumgartner R (1992) A biotinidase Km variant causing late onset bilateral optic neuropathy. Arch Dis Child 67:115–119. https://doi.org/10.1136/adc.67.1.115
Suormala T, Wick H, Baumgartner R (1988) Low biotinidase activity in plasma of some preterm infants: possible source of false-positive screening results. Eur J Pediatr 147:478–480. https://doi.org/10.1007/BF00441970
Vallejo-Torres L, Castilla I, Couce L, Pérez-Cerdá C, Martín-Hernández E, Pineda M et al (2015) Cost-effectiveness analysis of a national newborn screening program for biotinidase deficiency. Pediatrics 136:424–432. https://doi.org/10.1542/peds.2014-3399
Canda E, Kalkan Uçar S, Çoker M (2020) Biotinidase deficiency: prevalence, impact and management strategies. Pediatric Health Med Ther 11:127–133. https://doi.org/10.2147/PHMT.S198656
From available databases of biotinidase variants are accessible online: the Leiden Open Variation Database; (https://grenada.lumc.nl/LOVD2/shared1/home.php?select_db=BTD).
From available databases of biotinidase variants are accessible online: ClinVar; https://www.ncbi.nlm.nih.gov/clinvar/
From available databases of biotinidase variants are accessible online: “Biotinidase Deficiency and BTD,” hosted by ARUP Laboratories (http://www.arup.utah.edu/database/BTD/BTD_welcome.php).
From available databases of biotinidase variants are accessible online: https://varsome.com/gene/hg38/btd
From available databases of biotinidase variants are accessible online: https://www.mutationtaster.org/
From available databases of biotinidase variants are accessible online:https://www.ensembl.org/Homo_sapiens/Gene/Summary?g=ENSG00000169814;r=3:15601341-15722311
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, ACMG Laboratory Quality Assurance Committee (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–424. https://doi.org/10.1038/gim.2015.30
Taciane B, Fernanda L, Samyra L, Maria S, Carvalho R, Fonseca A et al (2017) Biotinidase deficiency: Genotype-biochemical phenotype association in Brazilian patients. PLoS One PLoS One 12:e0177503. https://doi.org/10.1371/journal.pone.0177503.Erratum.In:PLoSOne.2017;12:e0180463
Wiltink C, Kruijshaar E, van Minkelen R, Onkenhout W, Verheijen W, Kemper A et al (2016) Neonatal screening for profound biotinidase deficiency in the Netherlands: consequences and considerations. Eur J Hum Genet 24:1424–1429. https://doi.org/10.1038/ejhg.2016.65
Karaca M, Özgül K, Ünal Ö, Yücel D, Kılıç M, Hişmi B et al (2015) Detection of biotinidase gene pathogenic variants in Turkish patients ascertained by newborn and family screening. Eur J Pediatr 174:1077–1084. https://doi.org/10.1007/s00431-015-2509-5
Seker B, Mungan O, Kor D, Bulut D, Seydaoglu G, Öktem M et al (2018) Twenty-seven pathogenic variants with three novel pathologenic variants causing biotinidase deficiency: a report of 203 patients from the southeastern part of Turkey. J Pediatr Endocrinol Metab 31:339–343. https://doi.org/10.1515/jpem-2017-0406
Canda E, Yazici H, Er E, Kose M, Basol G, Onay H et al (2018) Single center experience of biotinidase deficiency: 259 patients and six novel pathogenic variants. J Pediatr Endocrinol Metab 31:917–926. https://doi.org/10.1515/jpem-2018-0148
Schulpis H, Gavrili S, Tjamouranis J, Karikas A, Kapiki A, Costalos C (2003) The effect of neonatal jaundice on biotinidase activity. Early Hum Dev 72:15–24
Cowan M, Blitzer G, Wolf B, Working Group of the American College of Medical Genetics Laboratory Quality Assurance Committee (2010) Technical standards and guidelines for the diagnosis of biotinidase deficiency. Genet Med 12:464–470
Swango L, Demirkol M, Hüner G, Pronicka E, Sykut-Cegielska J, Schulze A et al (1998) Partial biotinidase deficiency is usually due to the D444H pathogenic variant in the biotinidase gene. Hum Genet 102:571–575
Norrgard J, Pomponio J, Swango L, Hymes J, Reynolds T, Buck A, Wolf B (1998) Double mutation (A171T and D444H) is a common cause of profound biotinidase deficiency in children ascertained by newborn screening the the United States. Hum Mutat 11:410
Procter M, Wolf B, Mao R (2016) Forty-eight novel mutations causing biotinidase deficiency. Mol Genet Metab 117:369–372. https://doi.org/10.1016/j.ymgme.2016.01.002
Mühl A, Möslinger D, Item CB, Stöckler-Ipsiroglu S (2001) Molecular characterisation of 34 patients with biotinidase deficiency ascertained by newborn screening and family investigation. Eur J Hum Genet 9:237–243
Hsu H, Chien H, Hwu L, Chang F, Ho C, Chou P et al (2019) Genotypic and phenotypic correlations of biotinidase deficiency in the Chinese population. Orphanet J Rare Dis 14:6. https://doi.org/10.1186/s13023-018-0992-2
Sivri H, Genç A, Tokatli A, Dursun A, Coşkun T, Aydin I et al (2007) Hearing loss in biotinidase deficiency: genotype-phenotype correlation. J Pediatr 150:439–442. https://doi.org/10.1016/j.jpeds.2007.01.036
Baykal T, Gokcay G, Gokdemir Y, Demir F, Seckin Y, Demirkol M et al (2005) Asymptomatic adults and older siblings with biotinidase deficiency ascertained by family studies of index cases. J Inherit Metab Dis 28:903–912
Acknowledgements
We would like to thank Dr. Mertcan Karaoglan for his contributions and suggestions for the fluent writing of the article in English.
Funding
No funding.
Author information
Authors and Affiliations
Contributions
Study conception and design were done by Karaoglan M and Nacarkahya G; data were collected by Karaoglan M and Nacarkahya G. Interpretation of results was done by Karaoglan M, Nacarkahya G, Aytaç EH, and Keskin M. Draft manuscript preparation was done by Karaoglan M, Nacarkahya G, and Aytaç EH. All authors reviewed the results and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Authors declare to adherence to the international ethical standards. All the procedures carried out in the research with participation of humans were in compliance with the ethical standards of the National Research Ethics Committee and with the Helsinki Declaration of 1964 and its subsequent changes or with comparable ethics standards. The study was approved by the Gaziantep University Clinical Research Ethics Committee. Decision no/date: 2019/194–19.06.2019. Informed voluntary consent was obtained from every participant of the study.
Competing interests
All authors have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Karaoglan, M., Nacarkahya, G., Aytac, E.H. et al. Genotype-biochemical phenotype analysis in newborns with biotinidase deficiency in Southeastern Anatolia. Egypt J Med Hum Genet 25, 28 (2024). https://doi.org/10.1186/s43042-024-00500-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-024-00500-x