
Abstract
Background
LHPP is a tumor suppressor protein associated with various malignancies like liver, oral, pharyngeal, bladder, cervical, and gastric cancers through controlling various pathways. Several single nucleotide variants have been reported to cause cancers. The main objectives of our study were to investigate the impact of the deleterious non-synonymous single nucleotide variants on structure and functions of the LHPP protein.
Results
We used nine computational tools (SNAP2, PROVEAN, POLYPHEN 2, PREDICT SNP, MAPP, PhD-SNP, SIFT, PANTHER, and PMUT) to find out the deleterious SNPs. These nine computational algorithms predicted 34 nsSNPs to be deleterious as a result of their computational analysis. Using ConSurf, I-Mutant, SDM, MUpro, and Mutpred, we emphasized more how those harmful nsSNPs negatively affect the structure and function of the LHPP protein. Furthermore, we predicted the mutant protein structures and assessed the total energy value deviation in comparison with LHPP original structure and also calculated RMSD values and TM scores. By comparing the result from all these computational approaches, we shortlisted a total eight novel nsSNPs (D214G, D219N, Q224P, L231P, G236W, R234C, R234P, and V233G) that impose high risks to the structure and functions of LHPP protein. To analyze the mutant protein’s behavior in physiological condition, we performed 50 ns molecular dynamic simulation using WebGro online tool and found that the mutants values vary from the wild type in terms of RMSD, RMSF, Rg, SASA, and H-bond numbers. Prognostic significance analysis by Kaplan–Meier plotter showed that abnormal regulation of LHPP can also serve as a prognostic marker for the patient with breast, ovarian, and gastric cancers. Additionally, ligand binding sites analysis revealed the presence of D214G and D219N mutants in the binding site one which means these two nsSNPs can disturb the binding capacity of the LHPP protein. Protein–protein interaction analysis revealed LHPP proteins’ interactions with PPA1, ATP12A, ATP4A, ATP4B, ATP5F1, ATP5J, PPA2, ATP6V0A4, ATP6V0A2, and MT-ATP8 with different degree of connectivity.
Conclusion
These results demonstrate a computational understanding of the harmful effect of nsSNPs in LHPP, which may be useful for molecular approaches.
Similar content being viewed by others

Background
As a member of halo-acid dehalogenase like hydrolase domain-containing (HDHD) gene family [1], phosphohistidine phosphate inorganic pyrophosphatase (LHPP) inhibits the growth of tumors in a variety of human organs [2]. The protein coded by the human LHPP gene (NM 022126) which can be found on chromosome 10q26.13 [3] weighs 29 KD and consists of 270 amino acid [4]. It is a non-transmembrane, hydrophobic, and non-secretory protein, which is encyclopedically expressed mainly in the cytoplasm in most tissues such as urinary bladder, kidney, liver, and brain [4,5,6,7]. This LHPP protein consists of three leucine zipper domains and from worms to humans, it is a highly evolutionary preserved histidine phosphatase that was first discovered in swine brain tissues [2]. This gene is mainly expressed in thyroid and brain tissue [8].
It is believed that LHPP may be directly linked to the development of several types of cancer in humans, such as bladder cancer, liver cancer, oral and pharyngeal cancers, cervical cancer, and gastric cancer [9] (Table 1). Low level of LHPP expression is closely linked with an rise in tumor malignancy and a reduction in overall survival [1]. Recent studies have found that tumorigenesis in stomach, breast, esophageal, skin, head and neck, bladder, lung, liver, and pancreas tissues is mainly caused by LHPP mutation and its decreased expression. LHPP mutations were found in a wide range of tumors [9]. Forty nine LHPP mutations involvement were acknowledged in a search through the TCGA and the international cancer genome consortium (ICGC) databases in several human cancers, for example, liver, skin, breast, bladder, stomach, head and neck, esophagus cancers [9]. Moreover, LHPP is also crucial to prevent stress-related illness like depression [10].
The emergence of a variety of cancers is mediated by various signaling pathways. The PI3K/AKT/mTOR signaling cascades play a role, either directly or indirectly, in the progression of various types of malignant tumors [18]. Through controlling the PI3K/AKT signaling pathway, LHPP influences the development of human malignancies. It can prevent cancer cells from proliferating and metastasizing while also promoting their apoptosis through the regulation of AKT as it is reported to suppress the expression of p53. The biological signaling system of phosphatidylinositol 3-kinase and protein kinase B (PI3K/AKT) is crucial in controlling the propagation, apoptosis, and development of cancer cells [1]. The phosphorylation of PI3K/AKT is significantly lowered by the overexpression of LHPP proteins, which prevents tumor progression.
Furthermore, LHPP performs as a restrainer of the Wnt/catenin signaling pathway. When LHPP is overexpressed, it lowers phospho-GSK-3 levels and depletes active catenin, which in turn inactivates Wnt/catenin signaling [3]. Furthermore, LHPP inhibits Wnt/-catenin signaling via controlling Akt activation [3], where LHPP up-regulation lowers p-Akt levels and influences Akt's downstream targets [19]. Besides, Akt serves as a crucial upstream regulator of GSK-3, and when it is activated, it phosphorylates GSK-3, causing GSK-3 to become inactive and Wnt/-catenin signaling to become active [20].
Single nucleotide polymorphisms (SNPs) are thought to be the most prevalent form of variations in DNA sequence, account for the majority (90%) of genetic variants in the human genome [21]. Non-synonymous SNPs (nsSNPs), alternatively referred to as missense SNPs, hold great importance as they result in substitutions of amino acid residues, causing functional diversity in human proteins. This variation either can be neutral or deleterious. Potentially negative effects such as protein structure destabilization, changes in gene regulation, and influences on protein charge, shape, hydrophobicity, firmness, dynamics, translation, and inter/intra protein connections compromise the structural integrity of cells. This non-synonymous SNPs can also modify DNA and transcriptional binding factors, retaining the structural integrity of cells and tissues while having an impact on gene regulation [22]. Past research has demonstrated that around 50% of mutations associated in various genetic illnesses are caused by nsSNPs [23, 24]. Recently, the structural and functional effects of nsSNPs on different tumor suppressor proteins of the human genome have been predicted by numerous studies using in silico analysis [25,26,27].
Several nsSNPs have been found in LHPP that are responsible for LHPP gene’s impeded activity which eventually lead to tumorigenesis. LHPP (rs201982221) is found to be linked with oral cavity and pharyngeal cancers in a genome-wide study [28]. Besides, by suppressing the expression of Cyclin B1, Pyruvate Kinase M2, and Matrix Metallo Proteinase 7/9 (CCNB1, PKM2, and MMP7/9), high LHPP expression reduces the proliferation and spread of various human cancer cells [12]. Given the significance of LHPP in human health and sickness, the primary goals of this analysis were to identify the most harmful missense SNPs and to assess how the SNPs affect the protein's structure and functions. So far, there hasn't been any significant in silico study on LHPP protein that analyzes the impact of SNPs on both sequencing and structural properties except Mahmood et al. predicted the association of L22P, I212T, G227R, and G236R nsSNPs with hepatocellular carcinoma [11]. Taking into consideration of LHPP proteins’ role in various cancer types (Table 1) with rising data, we have carried out extensive analysis and used a variety of bioinformatics tools to find novel single nucleotide variants that impose high risks to the function and structure of LHPP protein in various cancer types. An outline of the whole procedural tactics is summarized in the following schematic diagram (Fig. 1).

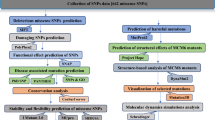
Overall strategies employed in this study. Following nsSNPs retrieval from the dbSNP database, deleterious nsSNPs were identified using nine different computational tools. Selected SNPs were tested whether affect proteins’ stability or not using I Mutant, SDM, and MuPro tools. After that, evolutionary conservation of the deleterious SNPs was predicted using ConSurf followed by their functional and structural modifications identification using MutPred. Structural effects of point mutation were observed and then cancer-associated SNPs were identified. Then, different cancer patients’ survival analysis was performed using Kaplan–Meier plotter followed by ligand binding sites prediction using FTSite tool. LHPP protein–protein interaction network was predicted using the STRING database. At last, a 50 ns simulation was carried out using WebGro tool to assess the mutant structures’ stability in terms of RMSD, RMSF, Rg, SASA, and Hydrogen bonds values
Methods
Retrieval of nsSNPs
The NCBI dbSNP database, which is the largest SNP database, was used to get the desired LHPP gene SNPs [29, 30] (https://www.ncbi.nlm.nih.gov/snp/). SNPs have been categorized into nine main groupings based on where they are present in the genome: synonymous, frameshift, in frame deletion, in frame indel, in frame insertion, initiator codon variation, intron, and missense [31]. This site provided information on missense SNPs (SNP ID), protein accession numbers, positions, and residue changes. The RCSB PDB was used to obtain the protein structural file [32].
Screening of deleterious nsSNPs
The influence of missense single nucleotide polymorphisms (SNPs) on the LHPP gene's structure and function was anticipated using a variety of computational programs. To evaluate the most harmful nsSNPs, we made use of nine different in silico nsSNP prediction techniques (SNAP2, PROVEAN, POLYPHEN 2, PREDICT SNP, MAPP, PhD-SNP, SIFT, PANTHER, and PMUT).
SNAP2 (https://www.rostlab.org/services/snap/) uses only sequence-based computationally acquired data, and using (screening for non-acceptable polymorphisms) this it is able to categorize all nsSNPs in all proteins as harmful (impact on function) or neutral (no effect). SNAP assigns a reliability score to each occurrence, which serves as a well-calibrated indicator of the degree of confidence in a specific prediction [33]. SNAP2 enables a rapid evaluation of functionally important positions in new proteins and offers a reliable prediction of variant effects [34].
The PROVEAN (http://provean.jcvi.org/index.php) enables high-throughput genomic and protein variant analysis for human and mouse variants. It provides a fast analysis of protein variations from any organism. For large sets of mouse and human genome-wide nucleotide or amino acid variants, this web interface can now offer a precomputed forecast. PROVEAN's cutoff was set at -2.5 for well-balanced accuracy where amino acid alterations score > − 2.5 is regarded as a deleterious mutation. Researchers can, however, reapply their individual cutoffs to their investigation to improve either sensitivity or specificity [35].
POLYPHEN 2 (polymorphism phenotyping v2) (http://genetics.bwh.harvard.edu/pph2/) assesses the possible effect of amino acid dissimilarities on the stability and functionality of human proteins using structural and comparative evolutionary factors. A number of sequences, phylogenetic, and structural characteristics that define the substitution provide the foundation of the prediction [36]. The PolyPhen-2 value ranges from 0 (tolerated) to 1 (deleterious). Variants predicted to be benign have scores of 0. More confidently anticipated to have negative effects are values closer to 1.0. For the query submission, the FASTA sequence of a protein and information about amino acid substitution are needed [37].
The PredictSNP (https://loschmidt.chemi.muni.cz/predictsnp/) is a consensus classifier that combines the eight top prediction techniques (MAPP, nsSNPAnalyzer, PANTHER, PhD-SNP, PolyPhen-1, PolyPhen-2, SIFT, and SNAP) to give a more reliable and alternate prediction that is accurate made by individual integrated programs. A FASTA formatted amino acid sequence of a query protein and the mutations are used as input. The PredictSNP score's fall within the continuous range of < − 1, + 1 > . The mutations are regarded as neutral, if the score is between − 1 and 0 and deleterious for the values in the interval (0, + 1 >) [38].
MAPP (multivariate analysis of protein polymorphism) forecasts the functional impact of altered amino acids on the basis of the evaluation of physicochemical properties discovered in the protein sequence alignment. The likelihood that amino acid replacement will impair the protein's ability to function normally increases with the computed deviation. MAPP's interpretability of its impact ratings, which offer a transparent justification of predictions in terms of physicochemical features, complements its ability to forecast outcomes accurately [39].
PhD-SNP (https://snps.biofold.org/phd-snp/) is intended to be lightweight and simple which is only trained on comparative data in the form of the conservation score derived from numerous sequence alignments. The purpose of this tool is to ascertain whether a specific single-point protein mutation is a benign polymorphism or linked to a pathology. An output from the PhD-SNP ranges from 0 to 1. When the score is greater than 0.5, pathogenic SNVs are expected; otherwise, it is benign [39].
SIFT (sorting intolerant from tolerant) (https://sift.bii.a-star.edu.sg/) employs sequence homology to determine if an amino acid replacement will have an impact on protein function and, perhaps, change phenotype. SIFT delivers a prediction score against submitted rsID for the nsSNP query, with a prediction score of 0.05 or higher being considered intolerant and 0.05 or lower being considered tolerant [40]. The SIFT algorithm just uses the sequence to make predictions.
PANTHER (protein analysis through evolutionary relationships) (http://www.pantherdb.org/) uses family multiple sequence alignments and phylogenetic trees to estimate whether a query protein amino acid alteration would likely affect protein function [41, 42]. If protein sequences coupled with human missense variations are supplied as a query, PANTHER offers position-specific evolutionary conservation values [43].
PMUT (http://mmb.irbbarcelona.org/PMut/analyses/new/) enables the quick and precise prediction (80% success rate in humans) of the pathogenic character of single-point amino acids changes based on the usage of neural networks. The pathogenicity score ranges from 0 to 1. A score higher than 0.5 signals pathological mutations. The PMUT server exhibits the location of the mutation on the protein structure, utilizing a color-coding system to indicate the pathogenicity of the mutation. The software has the capability to simulate numerous single-point mutations throughout the entire sequence, aiding in the identification of regions where mutations are anticipated to have a substantial pathological effect [44].
Analyzing protein stability
To check the stability of the target protein, I Mutant, SDM, and MuPro tools were used. It is possible to determine with the aid of I Mutant (http://gpcr2.biocomp.unibo.it/cgi/predictors/I-Mutant3.0/I-Mutant3.0.cgi) whether or not a change in amino acid in a protein sequence will or won't affect the firmness of the protein. The tool makes use of information from ProTherm, the largest experimental database on protein mutations. I- Mutant2.0 facilitates the prediction of alterations in protein stability across different temperature and pH ranges. It forecasts the reliability index (RI) of the results on a scale of 0–10, with 10 being the most reliable [45, 46].
SDM is available at http://www-cryst.bioc.cam.ac.uk/sdm/sdm.php. A statistical potential energy function called the site directed mutator was created to forecast how SNPs will affect protein stability. Site-directed mutagenesis (SDM) can assist in directing the design of experiments or determining if a mutation will impact protein structure and contribute to disease. The input section must provide a wild-type structure as well as the position and type of the mutated amino acids [47].
MUpro (https://www.ics.uci.edu/~baldig/mutation.html) includes SVM and neural networks, two machine learning programs. The result predicts only whether or not the change will cause destabilization, without providing an actual ddG value. The user can choose from three different sorts of prediction outcomes when using MUpro, which employs protein sequence as its input information [48].
ConSurf's prediction of the evolutionary conservation of the most damaging missense nsSNPs
Critical locations in the concerned molecules can be found using ConSurf analysis (https://consurf.tau.ac.il/). It uses both the empirical Bayesian and maximum likelihood paradigms to calculate rate of evolution at each site. Based on phylogenetic relationships between homologous sequences, the ConSurf server calculates the evolutionary conservation of amino/nucleic acid positions in a protein/DNA/RNA molecule. ConSurf analyzes the phylogenetic relationship, performs multiple sequence alignment and sequence homology of the protein to determine the conservation of an amino acid in a protein. The scores range from 1 to 4 as a variable, 5–6 as intermediate, and 7–9 as conserved. The program also forecasts whether a certain residue will be buried or exposed, which can further highlight the residue's structural and functional significance [49].
Identification of functional and structural modifications
MutPred (http://mutpred.mutdb.org/) is an experimental study of phenotype-altering variations that is guided by probabilistic modeling of variant influence on specific characteristics of protein structure and function. Utilizing three separate built-in tools, namely Psi-BLAST, SIFT, and PFAM, which cover protein structure, function, and evolution, it categorizes a variant as disease-associated (pathogenic) or neutral. MutPred compares the structural and functional features of mutant and wild-type sequences. These variations are expressed as probabilities of structure and function gain or loss. A missense mutation with a MutPred score > 0.5 may be deemed "harmful," while one with a MutPred score > 0.75 should be regarded as having a high probability of being "damaging" [50].
Effect of point mutation on protein structure
Project HOPE (https://www3.cmbi.umcn.nl/hope/) analyzes the effects of amino acid substitutions on the native structures, as well as the hydrophobicity, charge, and size differences between wildtype and mutant residues. When a FASTA sequence or a Uniprot id is submitted as a query file, 3D homology modeling using the YASARA program yields significant findings about structural differences between mutant and native residues [51].
The energy minimization of a protein is calculated using Swiss PDB (Swiss PDB Viewer-Home (unil.ch)) [52] Viewer for various amino acid substitutions. It is a tool for modeling, visualizing, and analyzing protein structures. Structural data are retrieved directly using PDB structure. Energy minimization of a 3D structure of a protein is performed by this server using GROMACS program as a default force field that is built on the methods of steepest descent, conjugate gradient, and LBFGS (Limited-memory Broyden–Fletcher–Goldfarb–Shanno) algorithm [53].
Identification of cancer-associated nsSNPs
Mutation 3D (http://www.mutation3d.org/) is used to find clusters of amino acid substitution that arise from somatic cancer mutations. It is a useful tool for investigating the geographical distribution of amino acid changes on protein models and structures. When a target protein together with its mutations is inserted as a query, this program uses a 3D clustering approach to identify amino acid substitution of a protein that can cause cancer [54].
Structure analysis of wild-type and mutant models
The 3D structure of native protein was downloaded from PDB (2X4D), and models for mutant proteins were generated using PHYRE2 and SWISS-MODEL.
Phyre 2 uses cutting-edge distant homology identification techniques to create 3D protein models and analyze the effect of different amino acid combinations on a protein's structure and function. PDB file 2X4D was used as template, and the development of the mutant 3D structures of the LHPP protein was performed using Phyre 2 [55]. These models was visualized by Biovia Discovery Studio which is also used for sequence alignment, analyzing protein, and modeling data [56].
After that, utilizing TM-align [57] tool, comparative analysis of the structures of wild-type and mutant proteins was achieved. The TM-score is a number between 0 and 1, where 1 indicates that two structures perfectly match one another. The RMSD value associated with the mutant residues after superimposition with the natural protein structure was computed using PyMol, an open-source application for structural research [58].
The SWISS-MODEL server is used for the structural analysis of native and mutant structure using FASTA sequence of LHPP protein as input. When evaluating the model quality, the QMEAN scoring function is used to confirm the accuracy of the final models for both wild-type and mutant proteins. It also calculates z score ranging from 0 to 1 where 0 indicates a good match between model and experimental structures [52].
Survival analysis
Kaplan–Meier plotter (http://kmplot.com/analysis/index.php?p=background) was used to examine the prognostic value of LHPP gene expression for breast, ovarian, lung, and gastric cancers [59]. This database utilizes the Gene Expression Omnibus (GEO), European Phenome Atlas (EGA), and the Cancer Genome Atlas (TCGA) datasets for the data on overall survival rate and relapse-free rates that are available for cancer patients, meta-analysis-based biomarker discovery and evaluation [60]. Hazard ratio with 95% confidence intervals and logrank p-value were calculated and shown on the plot. The analysis was done in two categories of people according to the median expression of a gene. Through the use of microarray gene expression data from 21 different types of cancer, this algorithm can examine the potential effects of 54,675 genes (mRNA, miRNA, and protein) on the survival of 13,316 cancer patients, including 6235 breast, 3452 lung, 1440 gastric, and 2190 ovarian cancers [61].
Binding site prediction
FT site was used to predict the LHPP protein's ligand binding sites. Over 94% of apo-proteins have their binding sites predicted by FTSite, a service that also offers protein engineering, structure-based protein prediction, medication design, and an understanding of how proteins work together [62].
Protein–protein interaction
A protein's structure may change as a result of a mutation, which may also alter the protein's functionality. Consequently, interactions between mutant proteins and other proteins can have phenotypic implications. The STRING server [63] (https://string-db.org/) was used to examine how LHPP interacts with different proteins.
Molecular dynamics simulation analysis
An effective approach for studying the evolution of molecular systems and predicting their attributes from the underlying interactions is molecular dynamics (MD). The simulation for predicted structures were performed using WebGro server (https://simlab.uams.edu/) to check stability and flexibility [64]. A simple point charge (SPC) water model in a triclinic periodic box was used to solve the complex system and GROMOS96 43a1 force field settings was used. The temperature and pressure were set to, respectively, 300 k and 1.0 bar. There were 1000 frames per simulation, which took 50 ns to complete. The root mean square deviation (RMSD) of each atom and root mean square fluctuation (RMSF) of each amino acid residue were used to analyze the simulation's findings. We also perform H bond, radius of gyration (Rg) and solvent accessible surface area (SASA) analysis to investigate the effect of mutation.
Results
Retrieval of nsSNPs
The polymorphism information for the LHPP gene was collected using the NCBI dbSNP database. There were a total of 60,891 SNPs of which 1570 were noncoding transcript variants, 192 were synonymous, 59,393 were in the intron region, 421 were missense, and the rests were of other kinds. Since some reference SNP ID (rsID) contains multiple SNPs at a single site, a total of 323 missense variants were considered for our further study.
Identification of damaging nsSNPs
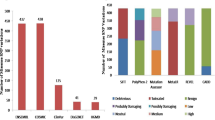
Nine distinct harmful SNP prediction tools, namely SNAP2, PROVEAN, POLYPHEN 2, PREDICT SNP, MAPP, PhD-SNP, SIFT, PMUT, and PANTHER were utilized to detect deleterious nsSNPs that can modify the structure or function of LHPP protein. Out of 323 nsSNPs, 35 nsSNPs were predicted to be deleterious (Table 2) by all nine in silico tools.
Prediction of changing structural stability
By using I-Mutant, SDM, and MUpro, which all perform tasks by taking into account single-site mutations, we were able to forecast any stability changes in the LHPP protein. The calculations were performed by I-Mutant using the reliability index (RI) value and free energy change values of ΔΔG and it predicted 31 nsSNPs decreased the stability of the protein, while 03 nsSNPs were found to increase the stability of the protein (Table 3). SDM tool predicted three nsSNPs (P190A, P190L, and P190R) as stabilizing and MuPro predicted P190L nsSNP as stabilizing substitution. We targeted only those nsSNPs which are predicted to be destabilizing by all the in silico tools for further analysis.
Evolutionary conservation analysis of deleterious nsSNPs in LHPP
The evolutionary conservation of amino acid residues of wild LHPP protein was predicted using the ConSurf server (Fig. 2). It was found that among 27 nsSNPs, 10 nsSNPs are in highly conserved regions of whom eight SNPs are buried and the rest two are exposed. Besides, Q224P is conserved and exposed while V186E, Y23D, Y23N, L22P, and I212T are also conserved but buried. Proteins are thought to be severely harmed by the nsSNPs (Additional file 1: Table S1) that are found in these conserved areas [23, 65].

Evolutionary conservation analysis of LHPP produced by ConSurf
Identification of functional and structural modifications
To identify the functional and structural modifications, the 27 nsSNPs that had been selected and determined to be harmful in the earlier steps were uploaded to the MutPred2 website and all of the nsSNPs showed a greater g value than 0.5. The structural and functional alterations predictions of these substitutions include altered ordered interface, altered stability, loss of relative solvent accessibility, altered DNA binding, altered metal binding, glycosylation and gain of phosphorylation, ubiquitination, and molecular recognition features.
The predicted data provided by this server showed that the various nsSNPs may be responsible for the structural and functional changes to the LHPP protein. All the SNPs (G227R, R234C, I212T, Y172S, L231P, L231R, L22P, N55T, V80G, Y23D, Y23N, D214G, V186E, G13R, G13W, R234P, G29C, D219N, V80G, G35D, and Q224P) exhibited very confident hypothesis with g value greater than 0.75 and p-value 0.05 (Additional file 1: Table S2). These estimated data offer convincing proof that these nsSNPs may have an impact on the structural and functional modifications of the LHPP protein.
Effect of point mutation on protein structure
To demonstrate how the physicochemical characteristics of wild-type and mutant amino acids differed in terms of size, charge, and hydrophobicity values, Project HOPE was used to create the 27 mutant LHPP protein 3D model structures (Additional file 1: Table S3). Apart from D219N, all of the mutant residues caused alteration in the size of the amino acid. G227R, L231R, G236R, G236W, R51S, V233M, V186E, G13R, G13W, L68F, and G35D are larger whereas R234C, I212T, Y172S, L231P, L22P, N55T, L91P, V80G, Y23N, Y23D, D214G, R234P (Fig. 3), G29C, Q224P, and V233G residues are smaller compared to the native structure. Besides, V233G, G35D, Y23D, Y23N, V80G, L231R, G29C, G227R, V186E, and I212T are less hydrophobic than wild type residue. And Y23D, Y23N, and I212T will cause loss of hydrophobic interactions in the core of the protein whereas V186E will cause loss of interaction in the surface of the protein. Furthermore, Q224P, R234P, R51S, G236W, G236R, N55T, G13W, and G13R are more hydrophobic and may result in possible loss of external interactions while D214G causes loss of H bond in the core of the protein and disturbs the correct folding. And in D219N, the charge of the buried wild-type residue is lost by this mutation (Fig. 3).

Structural effect of the point mutation on variant R234P (a and b) and D219N (c and d) predicted by HOPE server where green color indicates wild and red color indicates mutant residue
When an atom or molecule's position changes, Swiss PDB Viewer estimates the fluctuations in a protein's energy state. We identified the variations in the energy minimization state of the LHPP structure geometry between 27 variants and the wild-type protein (Table 4). The total amount of energy used by the native structure was − 20,422.141 kj/mol. G227R, I212T, L231R, Y23N, V233M, and G35D exhibited decrease in energy where rest of the variants showed increase in energy which is considered to be less favorable changes in comparison with native structure. After energy minimization, other missense variations indicated an increase in total energy. It was also found that G227R, L231P, L231R, L22P, G236R, G236W, L91P, D219N, Y23N, Y23D, V233M, V186E, G13R, G13W, R234P (Fig. 4), G29C, L68F, G35D, and Q224P exhibited both networks of clashes and H bond with nearby residue which was lacking in wild type residue. We have taken into consideration for our future studies those substitutions that showed an increase in energy following energy minimization.

Structural effect analysis by Swiss PDB Viewer. a represents R234 where four hydrogen bonds (green color) formed and b represents 234P where one clashes (pink color) found along with two hydrogen bonds after point mutation
Prediction of cancer-causing nsSNPs
This analysis is done by Mutation 3D server that predicts the harmful nsSNP that are associated with the development of cancer. Structural and functional changes in LHPP protein are due to mutation that results in tumor development. This analysis showed that D214G, D219N, Q224P, L231P, G236R, G236W, R234C, R234P, and V233G are associated with cancer. These nine cancer-associated nsSNPs are considered for further analysis. While these nine SNPs are the clustered mutation (colored red), SNPs (Y172S, L22P, N55T, L91P, R51S, V80G, Y23D, V186E, G13R, G13W, G29C, L68F) are covered mutation (colored blue) (Fig. 5).

Mutation 3D server predicted the association of nsSNPs (red mark) with cancer. Red color represents clustered mutation, while blue represents covered mutation. Nine SNPs are predicted to be associated with cancer
Structure analysis of wild-type and mutant models
PHYRE 2 and SWISS-MODEL computational tools were used to perform comparative structural analysis.
While Phyre 2 used LHPP proteins’ PDB file ‘2X4D’ as the template for predicting the 3D structures of the mutants, these structures were visualized by Biovia Discovery Studio, and further analysis of these structures were performed by calculating Tm-Score and RMSD values for each model (Table 5). The difference between the mutant structures and the wild type increases with increasing RMSD values. R234P displayed the highest RMSD value of 1.187 followed by L22P, Y23D, V233G, V186E and Q224P among other mutant models. Besides, nsSNPs R234C, G236R, and G236W showed a very slight deviation from the wild type model.
Furthermore, 3D structures of native LHPP protein and nine nsSNPs were analyzed using SWISS-MODEL to study the solvation and torsion with wild protein’s solvation score of 0.65 and torsion score of 1.03. L231P, G236R, G236W, D214G, R234P, Q224P, and V233G solvation scores are decreased. While the R234P torsion score is increased, D214G and D219N torsion scores remain the same. These physical modifications to the protein structure have therefore an impact on the side chains' capacity for molecular interaction (Additional file 1: Table S4).
Survival analysis
The prognostic significance of the LHPP gene expression for breast, ovarian, lung, and stomach cancers was examined using the Kaplan–Meier plotter by correlating gene expression and cancer patient survival. A log rank p-value and the hard ratio (HR) with 95% confidence intervals (CI) were computed. The plot analysis showed that LHPP dysregulation had various effects on various types of cancer. The HR ratio and p value for lung cancer were HR = 1.07 (0.94−1.21), log rank p = 0.29: the finding was not statistically significant. But in case of breast cancer and ovarian cancer, the LHPP gene had a HR and log rank p value HR = 0.85 (0.77−0.94); log rank p-value = 0.0019 and HR = 0.87 (0.76−0.99), p = 0.032, respectively, which are statistically significant and lower-level expression of LHPP is related to the less survival rate. Besides, in gastric cancer, the HR and p value were HR = 1.85 (1.51−2.28), log rank p-value = 2.5e−09 which is statistically very significant. This result showed that LHPP deregulation is associated with many different types of cancer and it can also serve as a prognostic marker for the patient with breast, ovarian and gastric cancers (Fig. 6).

Study of the relationship between LHPP protein expression and patient survival rates by means of Kaplan–Meier plotter that uses microarray-based data. Low level of LHPP expression is associated with the survival rates of patients with breast, ovarian, and gastric cancers
Ligand binding site prediction
Additionally, FTSite tool was used to reveal ligand binding sites of LHPP protein, which are shown in (Additional file 1: Table S5). It predicted three binding sites for LHPP whom all were visualized using the PyMol tool (Fig. 7). From this analysis, it was found that mutation positions of D214 and D219 are present in binding site one which can hamper its’ binding interactions with ligand.

a Ligand binding site of LHPP gene. First, second, and third binding sites are indicated by pink, green, and blue colors, respectively. b Ligand binding interaction at Asp(D) 214 in interaction site one
Protein–protein interaction analysis
For metabolite interaction and the maintenance of regular cellular activity, optimal protein–protein interaction is critical [66]. A protein's structure may change as a result of a mutation, which may also alter the proteins’ functionality. The STRING server was used to predict the interactions network of LHPP protein and it was found that LHPP protein has interactions with PPA1, ATP12A, ATP4A, ATP4B, ATP5F1, ATP5J, PPA2, ATP6V0A4, ATP6V0A2, and MT-ATP8 (Additional file 1: Table S6). Any change in LHPP protein can disrupt all these interactions (Fig. 8).

Protein–protein interaction network of LHPP protein predicted by STRING server
Molecular dynamic simulation analysis
To investigate the alteration of the mutant LHPP from its initial configuration under physiological circumstances, molecular dynamic simulation was performed. In order to examine the structural flexibility, stability, hydrogen bonding and solvency, the simulations were run for 50 ns.
Root mean square deviations (RMSD) data were used to calculate the total alterations in protein stability caused by the mutation. The native structure LHPP average RMSD is 0.0397 nm which was decreased in mutant D214G, Q224P, V233G, and R234P (Fig. 9a). The highest RMSD value was showed by mutant R234C with an average of ∼ 0.132 nm (Fig. 9b).

The RMSD values of wild LHPP protein (blue) and eight mutant proteins. Wild LHPP versus D214G, Q224P, V233G, R234P in (a). Wild LHPP versus D219N, L231P, R243C, G236W in (b)
To examine the structural flexibility, RMSF (root mean square fluctuation) analysis was performed. The highest residual fluctuation for native structure was 0.5409 nm and 0.3896 nm noticed at the position of Met 1 and Ser 241, respectively. Mutant L231P, R234C, Q224P, and D219N showed almost similar level of flexibility during 50 ns simulation. However, mutant V233G, G236W, D214G, R234P RMSF values varied greatly in comparison with wild structure. Among them, mutant R234P exhibited highest residual fluctuation (Fig. 10b). Overall, total RMSF value of all the mutant structures differed considerably from the native structure.

The RMSF values of wild LHPP protein (blue) and eight mutant proteins. Wild LHPP versus D214G, Q224P, V233G, R234P in (a). Wild LHPP versus D219N, L231P, R243C, G236W in (b)
In order to determine the stability of the protein, we also analyzed the total amount of hydrogen bonds present in the protein. The native structure exhibited an average of ∼ 337 H bonds while D219N, L231P, R234C, G236W, D214G, Q224P, V233G, and R234P exhibited, respectively, ∼ 201, ∼ 195, ∼ 194, ∼ 193, ∼ 199, ∼ 192, ∼ 196 and ∼ 192 H bonds throughout the 50 ns simulations (Fig. 11).

The number of hydrogen bonds present in wild LHPP protein and eight mutant proteins. Wild LHPP versus D219N, L231P, R243C, G236W in (a). Wild LHPP versus D214G, Q224P, V233G, R234P in (b)
Following that, we have also calculated solvent accessible surface area. The SASA (solvent accessible surface area) value of native and mutant proteins varies greatly during 50 ns simulations period. The native structure average SASA value was ∼ 0.90 nm2 and highest fluctuation was seen at the position of Leu 43. Mutant D219N, L231P, G236W, R234P exhibited similar SASA value to the native structure. However, R234C and Q224P showed greater and remaining mutant structure average SASA value was lower than the native structure (Fig. 12).

The SASA values of wild LHPP protein (blue) and eight mutant proteins. Wild LHPP versus D214G, Q224P, V233G, R234P in (a). Wild LHPP versus D219N, L231P, R243C, G236W in (b)
In order to evaluate the compactness and rigidity of the chosen proteins, the radius of gyration (Rg) was calculated. The Rg values of wild structure ranged from ∼ 1.82 nm to ∼ 1.89 nm. All the mutants exhibited fluctuation of Rg values compared to the wild one ranges from ∼ 1.80 nm to ∼ 1.9 nm (Fig. 13). D214G, Q224P, R234P, V233G, D219N, L231P, R234C, and G236W average Rg values were ∼ 1.88 nm, ∼ 1.85 nm, ∼ 1.83 nm, ∼ 1.87 nm, ∼ 1.87 nm, ∼ 1.87 nm, ∼ 1.84 nm, and ∼ 1.82 nm, respectively.

The Rg values of wild LHPP protein (blue) and its mutants). Wild LHPP versus D214G, Q224P, R234P, V233G in (a). Wild LHPP versus D219N, L231P, R243C, G236W in (b)
Fluctuation in RMSD, RMSF, Rg, H bonds, and SASA values indicating alteration in structures in mutant proteins.
Discussion
SNPs which are distributed widely throughout the human genome may have a significant impact on the structure and functionality of the relevant protein are crucial in the search of the causes of human diseases and traits, medication development, and human therapeutic responses [65, 67]. However, the abundance of SNPs presents a problem for scientists because it is costly, time-consuming, and labor-intensive to analyze every SNP using molecular methods to choose target SNPs. Applying in silico approaches may result in a better understanding of genetic dissimilarities in disease vulnerability and their phenotypic effects and a decrease in the number of candidates that need to be screened in molecular research [68].
In this study, we investigated the effect of point mutation on the structural and functional activity of phosphohistidine phosphate inorganic pyrophosphatase (LHPP) protein which has been involved in several numbers of cancers (Table 1). The human LHPP gene, positioned on chromosome 10q26.13, belongs to the HDHD gene family with three leucine zipper domains in it [2]. Thousands of polymorphisms in the LHPP genes’ coding and noncoding regions have already been identified.
Thirty-four missense SNPs were predicted deleterious by nine different computational tools amid 323 missense nsSNPs that have been identified to date. To investigate the effects of the identified thirty-four nsSNPs on protein stability, I-Mutant 3.0, SDM, and MuPro were utilized. The I-mutant tool projected that the G222R, L199P, and Q58P SNPs would increase the protein's stability whereas L101P showed no result. P190L, P190R, and P190A were predicted by the SDM tool to increase stability, but Q58P presented no result. And MuPro predicted P190L to increase the stability of the protein. Only those 27 SNPs were considered for further analysis which was forecast to decrease the stability of the protein by all three in silico tools. As changes in a protein's stability have an impact on its conformational shape, which controls how that protein functions, [67, 69], ConSurf conducted an investigation on the evolutionary conservation of chosen missense mutations to prioritize the identified most harmful SNPs for further analysis. The majority of the identified harmful SNPs were found at conserved positions and were structural and functional residues, which suggests that these SNPs may be harmful.
Using the MutPred2 web server, the causes of any molecular changes that might have an impact on the structure or function of the LHPP protein were investigated. Based on the g score and p score, all of the identified deleterious SNPs were "pathogenic" and were shown as potentially actionable, confident, and extremely confident hypothesis. Altered stability or altered ordered interfaces were the most consistently predicted effect in whole LHPP protein.
The Project Hope program offers in-depth knowledge of how point mutations harm a proteins’ structural configuration. While V233G, G35D, Y23D, Y23N, V80G, L231R, G29C, G227R, V186E, and I212T were less hydrophobic than wild type residue; in contrast, Q224P, R234P, R51S, G236W, G236R, N55T, G13W, and G13R were more hydrophobic which might cause the alteration of hydrophobic interactions. Additionally, D214G disrupts proper protein folding by causing a loss of the H bond in the protein's core. A change in an amino acid has an impact on the polar-polar interactions occurring within the protein molecule, which further alters the stabilization energy and destabilizes the protein [70]. The differences in stability were found by comparing particular characteristics of native and mutant structures. In the case of R234C, Y172S, L231P, L22P, N55T, G236R, G236W, L91P, R51S, D219N, V80G, Y23D, D214G, V186E, G13R, G13W, R234P, G29C, L68F, and Q224P, there were increased in energy which was calculated using Swiss PDB Viewer in comparison with the native structure. We targeted these variants for further analysis as these changes are considered less favorable. This shift in stability can be due to the variant residue's larger R group than the wild type, which cannot fit in the given space. Then, mutation 3D server analysis showed the association of D214G, D219N, Q224P, L231P, G236R, G236W, R234C, R234P, and V233G nsSNPs with cancer.
Understanding the overall effects of SNPs on proteins’ function depends heavily on the protein's 3D structure and conformation. As the 3D structure of the LHPP gene is already deposited in PDB, for the rest of the mutant 3D structures we have utilized Phyre 2, and SWISS-MODEL. The SWISS-MODEL analyzes solvation, and torsion value and it is known to impact protein stability as well as protein conformation and chain elasticity. These nine nsSNPs showed greater deviation in comparison with the native model. The structural effects of these mutations were examined by superimposing the wild-type and mutant protein models in PyMOL to calculate the RMSD value. The aberration between the two structures increased with increasing RMSD values, which predicted altered functional activity, and since all of these nine SNPs showed greater RMSD values which means it could be believed that these SNPs might greatly affect the function and structure of the LHPP protein.
To observe the prognostic characteristics, the Kaplan–Meier plotter bioinformatics tool was used and predicted that the LHPP gene dysregulation had a prognostic relevance and altered the overall survival rate of patients with ovarian, lung, and gastric cancers. In gastric and breast cancer, low expression is related to less survival rate. Any type of dysregulation caused by SNPs in the LHPP gene may have a significant impact on how long patients with breast and gastric cancer can survive. Furthermore, as the D214G and D219N mutations were found in the binding site in FT site analysis, the LHPP gene's ability to attach to its target may be affected.
It is also important to investigate how aberrant LHPP protein interacts with other interacting partner proteins to better understand the regulatory mechanism it uses. The result of the STRING analysis shows that the LHPP protein serves a variety of essential roles and connected with other proteins that may be involved in many pathways, and disruption of these pathways may lead to disorders. LHPP variants are also known to be associated with depressive disorder [71], risky sexual behavior, and alcohol dependence [72].
Our study finally predicted nine variants, namely D214G, D219N, Q224P, L231P, G236R, G236W, R234C, R234P and V233G associated with different types of cancer where G236R was also predicted to be associated with hepatocellular carcinoma in a research which also complements our study [11]. However, our study is not without limitations and was focused only on the coding region of the protein using a various number of computational algorithms and the PDB structure of the native protein was missing some side chain atoms.
To analyze the mutant protein’s behavior in the physiological condition, we performed molecular dynamic simulation using WebGro online tool and found that the mutants values vary from the wild type in terms of RMSD, RMSF, Rg, SASA, and H-bond numbers.
The outcomes of the MD simulations offer insightful information regarding the changes in native and mutant LHPP protein structures under physiological circumstances. The main focus of our analysis was mainly the deviation of mutant’s structure from the native structure. Only R234C showed higher RMSD value and on average mutant D214G, Q224P, R234P, D219N, L231P, and G236W displayed ∼ 0.30 nm, ∼ 0.26 nm, ∼ 0.27 nm, ∼ 0.29 nm, ∼ 0.31 nm, ∼ 0.27 nm, and ∼ 0.34 nm, respectively. These result indicates the higher structural deviation from the native structure as the lower RMSD value represents similarity to the target structure [73].
In order to analyze fluctuation caused by mutation, we also analyzed root mean square fluctuation (RMSF) value. We observed higher residual fluctuation from 235 to 244 residues for V233G, Q224P, D214G and for G236W residual fluctuation was spotted at 153–169 residues. In case of the native structure, the highest residual fluctuation was noticed at 1–10 aa and 244–255 aa.
Furthermore, from H bond analysis, we found that the number of H bonds in all mutant structure are lower than the native structure. Loss of H bond in mutant structure signified the loss of stability compared to the native structure. The SASA analysis also significantly illustrates the impact of mutation on LHPP structure as change in surface area may hamper ligand bindings, alter protein stability.
We calculated radius of gyration as well to evaluate the overall dimension of the protein (Fig. 13) exhibited that the mutant proteins were less compact in comparison with native structure suggesting structural changes due to mutation. The average value of native structure was ∼ 1.84 nm, and the mutants D214G, Q224P, R234P, V233G, D219N, L231P, R234C, and G236W average values were, respectively, ∼ 1.88 nm, ∼ 1.85 nm, ∼ 1.83 nm, ∼ 1.87 nm, ∼ 1.87 nm, ∼ 1.87 nm, ∼ 1.84 nm, and ∼ 1.82 nm. Therefore, it is evident that D214G, Q224P, V233G, D219N, L231P, and R234C mutants had higher radius of gyration compared to the native structure and it represents the lower stability compared to the native LHPP which ultimately leads to the functional dysfunction [74, 75].
It is clear that using computational approaches to quickly investigate the anticipated effects of variations remains a cost-effective strategy; besides, the more parameters that are considered, the more precise the forecast will be. By comparing the results of the above methods, we can conclude that these eight novel mutations (D214G, D219N, Q224P, L231P, G236W, R234C, R234P, and V233G) should be further confirmed through experimental approaches for their association with disordered LHPP function in addition to existing deleterious nsSNPs of this gene.
Conclusions
LHPP has been associated with several malignancies. Using computational prediction techniques, it was possible to determine the functional and structural effects of nsSNPs in the LHPP gene. Using SNAP2, PROVEAN, POLYPHEN 2, MutPred, PREDICT SNP, MAPP, PhD-SNP, SIFT, PANTHER, and PMUT, we were able to identify 35 harmful nsSNPs out of total 421. Among the 35, structural analysis showed that 27 SNPs had the greatest impact on the stability of LHPP protein. Further analysis identified eight nsSNPs with a high risk of being detrimental. In vitro and in vivo experimental studies can be designed by taking into account these in silico data and these findings will be valuable in the development of future therapeutic and diagnostic modalities.
Availability of data and materials
All data analyzed during this study are included in this article.
Abbreviations
- LHPP:
-
Phosphohistidine phosphate inorganic pyrophosphatase
- nsSNP:
-
Non-synonymous single nucleotide polymorphism
- PDB:
-
Protein data bank
- KD:
-
Kilodalton
- PPI:
-
Protein–protein interaction
- RMSD:
-
Root mean square deviation
- RMSF:
-
Root mean square fluctuation
- SASA:
-
Solvent accessible surface area
- Rg:
-
Radius of gyration
- H Bond:
-
Hydrogen bond
- MD:
-
Molecular dynamics
References
Wang D, Ning Z, Zhu Z et al (2021) LHPP suppresses tumorigenesis of intrahepatic cholangiocarcinoma by inhibiting the TGFβ/smad signaling pathway. Int J Biochem Cell Biol 132:105845
Yokoi F, Hiraishi H, Izuhara K (2003) Molecular cloning of a cDNA for the human phospholysine phosphohistidine inorganic pyrophosphate phosphatase. J Biochem 133(5):607–614
Li C, Yang J, Wang W et al (2021) LHPP exerts a tumor-inhibiting role in glioblastoma via the downregulation of Akt and Wnt/β-catenin signaling. J Bioenerg Biomembr 53(1):61–71
Lin J-X, Lian N-Z, Gao Y-X et al (2022) m6A methylation mediates LHPP acetylation as a tumour aerobic glycolysis suppressor to improve the prognosis of gastric cancer. Cell Death Dis 13(5):1–13
Makwana MV, Muimo R, Jackson RF (2018) Advances in development of new tools for the study of phosphohistidine. Lab Invest 98(3):291–303
Zhang J, Gelman IH, Katsuta E et al (2019) Glucose drives growth factor-independent esophageal cancer proliferation via phosphohistidine-focal adhesion kinase signaling. Cell Mol Gastroenterol Hepatol 8(1):37–60
Fuhs SR, Meisenhelder J, Aslanian A et al (2015) Monoclonal 1-and 3-phosphohistidine antibodies: new tools to study histidine phosphorylation. Cell 162(1):198–210
Wu F, Ma H, Wang X et al (2022) The histidine phosphatase LHPP: an emerging player in cancer. Cell Cycle 21(11):1140–1152
Liu S, Gao W, Lu Y et al (2022) As a novel tumor suppressor, LHPP promotes apoptosis by inhibiting the PI3K/AKT signaling pathway in oral squamous cell carcinoma. Int J Biol Sci 18(2):491
Lin D, Li L, Chen W-B et al (2023) LHPP, a risk factor for major depressive disorder, regulates stress-induced depression-like behaviors through its histidine phosphatase activity. Mol Psychiatry 28(2):908–918
Mahmood MS, Afzal M, Batool H et al (2022) Screening of pathogenic missense single nucleotide variants from LHPP gene associated with the hepatocellular carcinoma: an in silico approach. Bioinform Biol Insights 16:11779322221115548
Liao L, Duan D, Liu Y et al (2020) LHPP inhibits hepatocellular carcinoma cell growth and metastasis. Cell Cycle 19(14):1846–1854
Zheng J, Dai X, Chen H et al (2018) Down-regulation of LHPP in cervical cancer influences cell proliferation, metastasis and apoptosis by modulating AKT. Biochem Biophys Res Commun 503(2):1108–1114
Li Y, Zhang X, Zhou X et al (2019) LHPP suppresses bladder cancer cell proliferation and growth via inactivating AKT/p65 signaling pathway. Biosci Rep 39(7):BSR20182270
Sun W, Qian K, Guo K et al (2020) LHPP inhibits cell growth and migration and triggers autophagy in papillary thyroid cancer by regulating the AKT/AMPK/mTOR signaling pathway. Acta Biochim Biophys Sin 52(4):382–389
Wu F, Chen Y, Zhu J (2020) LHPP suppresses proliferation, migration, and invasion and promotes apoptosis in pancreatic cancer. Biosci Rep 40(3):BSR20184142
Hou B, Li W, Xia P et al (2021) LHPP suppresses colorectal cancer cell migration and invasion in vitro and in vivo by inhibiting Smad3 phosphorylation in the TGF-β pathway. Cell Death Discov 7(1):1–14
Feng Z (2010) p53 regulation of the IGF-1/AKT/mTOR pathways and the endosomal compartment. Cold Spring Harb Perspect Biol 2(2):a001057
Hindupur SK, Colombi M, Fuhs SR et al (2018) The protein histidine phosphatase LHPP is a tumour suppressor. Nature 555(7698):678–682
Hussain M, Xu C, Lu M et al (2017) Wnt/β-catenin signaling links embryonic lung development and asthmatic airway remodeling. Biochim et Biophys Acta BBA Mol Basis Dis 1863(12):3226–3242
Arshad M, Bhatti A, John P (2018) Identification and in silico analysis of functional SNPs of human TAGAP protein: a comprehensive study. PLoS ONE 13(1):e0188143
Rajasekaran R, Sudandiradoss C, Doss CGP et al (2007) Identification and in silico analysis of functional SNPs of the BRCA1 gene. Genomics 90(4):447–452
Kishimoto T, Ying B-W, Tsuru S et al (2015) Molecular clock of neutral mutations in a fitness-increasing evolutionary process. PLoS Genet 11(7):e1005392
Bernhofer M, Kloppmann E, Reeb J et al (2016) TMSEG: novel prediction of transmembrane helices. Proteins Struct Func Bioinf 84(11):1706–1716
Shantier SW, Elmansi HE, Elnnewery ME et al (2018) Computational analysis of single nucleotide polymorphisms (SNPs) in human TCell acute lymphocytic leukemia protein 1 (TAL1) gene/comprehensive study. bioRxiv. 447540.
Hossain M, Roy AS, Islam M (2020) In silico analysis predicting effects of deleterious SNPs of human RASSF5 gene on its structure and functions. Sci Rep 10(1):1–14
Pires AS, Porto WF, Franco OL et al (2017) In silico analyses of deleterious missense SNPs of human apolipoprotein E3. Sci Rep 7(1):1–9
Lesseur C, Diergaarde B, Olshan AF et al (2016) Genome-wide association analyses identify new susceptibility loci for oral cavity and pharyngeal cancer. Nat Genet 48(12):1544–1550
Bhagwat M (2010) Searching NCBI’s dbSNP database. Curr Prot Bioinf 32(1):1–19
Morales J, Pujar S, Loveland JE et al (2022) A joint NCBI and EMBL-EBI transcript set for clinical genomics and research. Nature 604(7905):310–315
Pruitt KD, Tatusova T, Maglott D (2005) NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. 33(suppl_1):D501–D504.
Johnson M, Zaretskaya I, Raytselis Y et al (2008) NCBI BLAST: a better web interface. Nucleic Acids Res 36(suppl_2):W5–W9
Bromberg Y, Rost B (2007) SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res 35(11):3823–3835
Hecht M, Bromberg Y, Rost B (2015) Better prediction of functional effects for sequence variants. BMC Genomics 16(8):1–12
Choi Y, Chan APJB (2015) PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31(16):2745–2747
Adzhubei I, Jordan DM, Sunyaev S (2013) Predicting functional effect of human missense mutations using PolyPhen-2. Curr Prot Hum Genet 76(1):7–20
Adzhubei IA, Schmidt S, Peshkin L et al (2010) A method and server for predicting damaging missense mutations. Nat Methods 7(4):248–249
Bendl J, Stourac J, Salanda O et al (2014) PredictSNP: robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput Biol 10(1):e1003440
Stone EA, Sidow A (2005) Physicochemical constraint violation by missense substitutions mediates impairment of protein function and disease severity. Genome Res 15(7):978–986
Ng PC, Henikoff S (2003) SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res 31(13):3812–3814
Tang H, Thomas PDJB (2016) PANTHER-PSEP: predicting disease-causing genetic variants using position-specific evolutionary preservation. Bioinformatics 32(14):2230–2232
Thomas PD, Ebert D, Muruganujan A et al (2022) PANTHER: making genome-scale phylogenetics accessible to all. Protein Sci 31(1):8–22
Mi H, Poudel S, Muruganujan A et al (2016) PANTHER version 10: expanded protein families and functions, and analysis tools. Nucleic Acids Res 44(D1):D336–D342
Ferrer-Costa C, Gelpí JL, Zamakola L et al (2005) PMUT: a web-based tool for the annotation of pathological mutations on proteins. Bioinformatics 21(14):3176–3178
Bava KA, Gromiha MM, Uedaira H et al (2004) ProTherm, version 4.0: thermodynamic database for proteins and mutants. Nucleic Acids Res 32(suppl_1):D120–D121
Capriotti E, Fariselli P, Casadio R (2005) I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res 33(suppl_2):W306–W310
Choi I, Nelson J, Peterson L et al (2019) Sdm: a scientific dataset delivery platform. In: 2019 15th International Conference on eScience (eScience): IEEE, pp 378–387.
Cheng J, Randall A, Baldi PJPS et al (2006) Prediction of protein stability changes for single-site mutations using support vector machines. Proteins Struct Func Bioinf 62(4):1125–1132
Rubin M, Ben-Tal N (2021) Using consurf to detect functionally important regions in RNA. Curr Prot 1(10):e270
Mort M, Sterne-Weiler T, Li B et al (2014) MutPred Splice: machine learning-based prediction of exonic variants that disrupt splicing. Genome Biol 15(1):1–20
Venselaar H, Te Beek TA, Kuipers RK et al (2010) Protein structure analysis of mutations causing inheritable diseases. e-Sci Approach Life Sci Friend Interfaces 11(1):1–10
Guex N, Peitsch M (1997) SWISS-MODEL and the Swiss-Pdb viewer: an environment for comparative protein modeling. Electrophoresis 18(15):2714–2723
Johansson MU, Zoete V, Michielin O et al (2012) Defining and searching for structural motifs using DeepView/Swiss-PdbViewer. BMC Bioinf 13(1):1–11
Meyer MJ, Lapcevic R, Romero AE et al (2016) Mutation3D: cancer gene prediction through atomic clustering of coding variants in the structural proteome. Hum Mutat 37(5):447–456
Kelley LA, Mezulis S, Yates CM et al (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10(6):845–858
Design L (2014) Pharmacophore and ligand-based design with Biovia Discovery Studio®.
Zhang Y, Skolnick J (2005) TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res 33(7):2302–2309
Delano WL (2002) Pymol: an open-source molecular graphics tool. CCP4 Newsl Protein Crystallogr 40(1):82–92
Plotter K (2018) What is the KM plotter
Lacny S, Wilson T, Clement F et al (2018) Kaplan–Meier survival analysis overestimates cumulative incidence of health-related events in competing risk settings: a meta-analysis. J Clin Epidemiol 93:25–35
Goel MK, Khanna P, Kishore J (2010) Understanding survival analysis: Kaplan–Meier estimate. Int J Ayurveda Res 1(4):274
Ngan C-H, Hall DR, Zerbe B et al (2012) FTSite: high accuracy detection of ligand binding sites on unbound protein structures. Bioinformatics 28(2):286–287
Szklarczyk D, Gable AL, Nastou KC et al (2021) The STRING database in 2021: customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res 49(D1):D605–D612
Abraham MJ, Murtola T, Schulz R et al (2015) GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1:19–25
Miller MP, Kumar S (2001) Understanding human disease mutations through the use of interspecific genetic variation. Hum Mol Genet 10(21):2319–2328
Xu Y, Wang H, Nussinov R et al (2013) Protein charge and mass contribute to the spatio-temporal dynamics of protein–protein interactions in a minimal proteome. Proteomics 13(8):1339–1351
Vignal A, Milan D, SanCristobal M et al (2002) A review on SNP and other types of molecular markers and their use in animal genetics. Genet Sel Evol 34(3):275–305
Dakal TC, Kala D, Dhiman G et al (2017) Predicting the functional consequences of non-synonymous single nucleotide polymorphisms in IL8 gene. Sci Rep 7(1):1–18
Kong L, Rupp B (2016) Structural Biology communications. Protein stability: a crystallographer’s perspective. Acta Crystallogr Sect F Struct Biol Commun 72(2):72
Yue P, Li Z, Moult J (2005) Loss of protein structure stability as a major causative factor in monogenic disease. J Mol Biol 353(2):459–473
Cui L, Wang F, Yin Z et al (2020) Effects of the LHPP gene polymorphism on the functional and structural changes of gray matter in major depressive disorder. Quant Imaging Med Surg 10(1):257
Polimanti R, Wang Q, Meda SA et al (2017) The interplay between risky sexual behaviors and alcohol dependence: genome-wide association and neuroimaging support for LHPP as a risk gene. Neuropsychopharmacology 42(3):598–605
Uzoeto HO, Cosmas S, Ajima JN et al (2022) Computer-aided molecular modeling and structural analysis of the human centromere protein–HIKM complex. Beni-Suef Univ J Basic Appl Sci 11(1):101
Odiba AS, Durojaye OA, Ezeonu IM et al (2022) A new variant of mutational and polymorphic signatures in the ERG11 gene of fluconazole-resistant candida albicans. Infect Drug Resist. 3111–3133.
Lobanov MY, Bogatyreva N, Galzitskaya O (2008) Radius of gyration as an indicator of protein structure compactness. Mol Biol 42:623–628
Acknowledgements
Not applicable.
Funding
There is no funding source for this research.
Author information
Authors and Affiliations
Contributions
MKI conceived and designed the experiments and made critical revisions. TF and MKI analyzed the data and contributed to the preparation of the manuscript. All authors approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
: Table S1. Evolutionary conservancy of amino acids in LHPP protein analyzed by Consurf server. Tables S2. LHPP proteins’ functional and structural alterations predicted by MutPred2 tool. Table S3. Structural effects of nsSNPs on LHPP protein using Project Hope. Table S4. Comparative impacts of nsSNPs on 3D structure of LHPP investigated by Swiss Model. Table S5. Binding site prediction by FT site server. Table S6. Functions of proteins connected with LHPP.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Feroz, T., Islam, M.K. A computational analysis reveals eight novel high-risk single nucleotide variants of human tumor suppressor LHPP gene. Egypt J Med Hum Genet 24, 47 (2023). https://doi.org/10.1186/s43042-023-00426-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-023-00426-w