
Abstract
Background
Cerebrotendinous xanthomatosis (CTX) is a rare autosomal recessive lipid storage disorder that leads to multisystem involvement. It is caused by mutations in the CYP27A1 gene which encodes the mitochondrial enzyme sterol 27-hydroxylase.
Case presentation
Herein we describe two affected CTX siblings with symptoms including seizures, severe diarrhea (steatorrhea), vomiting, and developmental motor delay, which was initially misdiagnosed as Short-Chain acyl-CoA dehydrogenase (SCAD) deficiency. However, to identify the possible genetic cause(s) of the disease, whole exome sequencing (WES) was performed. It was confirmed that these patients carried a nonsense variant (c.808C > T; p.Arg270Ter) of the CYP27A1 gene. The variant in the CYP27A1 gene was classified as pathogenic.
Conclusion
We report rare cases of CTX with a novel mutation and summarize the clinical and molecular pathogenesis of this disease. Genetic analysis should be used as the conclusive method for CTX diagnosis because of the multisystem involvement and the lack of specific symptoms. The variant in these patients expands the molecular and phenotypic basis of a variant in CTX.
Similar content being viewed by others


Background
Cerebrotendinous xanthomatosis (CTX: OMIM#213700) is a rare autosomal recessive bile acid biosynthesis disorder described in 1937. It is caused by sterol 27-hydroxylase deficiency (CYP27A1, EC 1.14.15.15), i.e., the mitochondrial cytochrome P 450 enzyme, due to mutations in the CYP27A1 gene [1]. The CYP27A1 gene spans 18.6 kb of DNA, which is located on chromosome 2q33-qter and contains nine exons and eight introns. A mature protein of 498 amino acids and 33-residue mitochondrial signal sequence constructs the sterol 27-hydroxylase enzyme, which maintains the binding site for heme and adrenodoxin. The prevalence of CTX is reported to be 3 to 5 per 100,000 but, it is probably underestimated [2, 3].
Regarding sterol 27-hydroxylase deficiency, the cholesterol and cholestanol metabolites accumulate in several tissues such as the central nervous system, muscle tendons, and eye lenses [4, 5]. Therefore, the clinical presentation of CTX is wide-ranging and highly heterogeneous, resulting in a difficult diagnosis, especially in the early stages. Broad systemic and neuropsychiatric clinical manifestations of CTX are reported including neonatal jaundice, cholestasis, juvenile bilateral cataracts, refractory diarrhea, osteoporosis, tendon xanthomas, seizures, developmental delay, intellectual disability, and progressive neuropsychiatric disturbances [6]. It is known that early diagnosis and treatment of chenodeoxycholic acid (CDCA), as a useful medicine, play a crucial role in the prevention of the development of neurological dysfunction [7].
In the present study, we report two CTX siblings with a delayed diagnosis who showed relative response after treatment with CDCA.
Case presentation
This study recruited an Iranian family with two affected children a girl (proband) and her sister aged 7.5 and 6 years, respectively. The parents’ marriage was consanguineous (first cousin). They enrolled in the database registry of Metabolic (no. 1400-7860), Shiraz University of Medical Sciences.
Case 1 The 7.5-year-old girl was proband and she was delivered following an uneventful, normal, and term pregnancy with a birth weight of 2.85 kg and height of 49 cm.
At age one, the proband initially reported seizures, severe diarrhea (steatorrhea), vomiting, and developmental motor delay. She had vomiting accompanied by seizures two times. Biochemical results showed a high level of lactate (27 mg/dl), homocysteine (21.7 μmol/L), total cholesterol (280 mg/dL), and triglycerides (164 mg/dL) (Table 1). A fecal fat test confirmed the presence of steatorrhea. Organic acids and acylglycines profile showed a normal amount of methylmalonic acid, methylsuccinic acid, and mildly increased butyrylglycine (4.3 mmol/mol creatinine). The acylcarnitine profile in the plasma by LC–MS/MS indicated that the ratio C4/C0 (0.027, normal: < 0.018) was mildly increased. The alanine aminotransferase (ALT), aspartate aminotransferase (AST), urine creatinine, prothrombin time (PT), partial thromboplastin time (PTT), sodium, potassium, fasting blood sugar (FBS), and BUN were normal. Neurological examination demonstrated cognitive decline, increased deep tendon reflexes as well as mild muscle hypertonia. Many cutaneous xanthomas on the back were also detected.
Based on the above results, the metabolic disorder specifically the short-chain Acyl-CoA dehydrogenase deficiency (SCAD), was suggested. Despite all types of treatment, she had a progressive problem with walking and speaking, and overall cognition. Regarding the progressive and deteriorating disease, at the age of 5 years, molecular analysis was suggested for confirmation of diagnosis. Whole exome sequencing (WES) was performed to analyze all exons of protein-coding genes as well as some important other genomic regions. Analyses were performed using an Illumina Hiseq4000 (Illumina Inc., San Diego, CA, USA). It uses a 100 bp paired-end read with a mean depth of coverage of 55 × with 95.5% and 91.5% coverage at 10 × and 20 × , respectively. The human genome 19 and the in-house database of 800 Iranian control samples were used as the reference. For in silico analysis, Polyphen2, scale-invariant feature transform (SIFT), and Mutation Taster were used. Genomic Evolutionary Rate Profiling (GERP) and Phastcons scores were used to evaluate the conservation of the variants. The variant interpretation was according to American College of Medical Genetics and Genomics (ACMG) guidelines [8]. Sanger sequencing was applied for validation of the identified variants using An ABI Prism 3500 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). WES results showed that the patient was homozygous for the c.808C > T (p.Arg270Ter) variant in exon 4 of the CYP27A1 gene [9]. In silico analysis revealed that the variant, c.808C > T (p.Arg270Ter) was pathogenic, which would have possibly resulted in premature termination of the protein (Fig. 1). In addition, Sanger sequencing analyzed the samples from the parents. It should be mentioned that both parents were asymptomatic.

Genetic analysis identified a CYP27A1 variant. A Pedigree of the family in the CYP27A1 variant carrier. Black arrow shows proband, B DNA chromatogram shows a homozygous C-to-T transition at nucleotide 808 of CYP27A1, predicting a substitution of an arginine for stop codon at residue 270 (p.Arg270Ter). + indicates variant positive, − indicates variant negative. (CYP27A1: NG_007959.1)
As a result, high-dose CDCA treatment (5 mg/kg/day 3 times per day) was initiated for the patient. After 4 months, her spinal cord xanthomas began to diminish, also improvement in her intellectual ability and movement was observed. However, because of non-compliance, CDCA treatment was repeatedly discontinued; CDCA treatment was stopped, diarrhea recurred, and cognitive abilities and walking worsened.
In the last follow-up, at the age of 7.5 years old, she still had difficulty walking and needed help to get up and climb the stairs. She also had speech problems and a learning disorder in school. However, other problems such as muscle hypertonia, and cognitive problems were diminished.
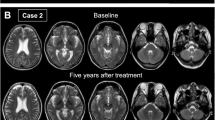
Case 2 A 6-year-old female was the youngest sister of the proband. She was born at 35 weeks of gestational age after a normal and uneventful pregnancy. Her symptoms and signs were very similar to her siblings including severe diarrhea (steatorrhea), vomiting, and motor delay. At the age of two, she had a high HDL level (71 mg/dL, normal range: 40–60) and aspartate aminotransferase (AST) (41 IU/L, normal range: 1–31). Like case 1, she was misdiagnosed as short-chain Acyl-CoA dehydrogenase deficiency (SCAD) and treated accordingly. After performing molecular analysis and WES, at the age of 3.5, she was diagnosed with CTX. She was carrying the same homozygous variant c.808C > T (p.Arg270Ter) in exon 4 of CYP27A1 gene as case 1. Treatment with a high dose of CDCA (5 mg/kg/day 3 times per day) started. After treatment, she showed a dramatic response and quick improvement and all symptoms disappeared in less than one month.
In the last follow-up, at the age of six, she had no problem in walking, speaking, cognition, and learning at school.
Discussion and conclusions
CTX is an inherited lipid storage disorder with a wide range of manifestations, which can show variable types, manifestations, and severity, even within the same family. The majority of common clinical presentations are chronic diarrhea, progressive neurologic dysfunction, cataract, and tendon xanthoma. However, the reported results of more than 300 patients so far have demonstrated that the diagnosis of CTX is still challenging because of varied clinical manifestations [10]. Herein, we are reporting a homozygous variant in the CYP27A1 gene, leading to the loss of function of sterol 27-hydroxylase, in patients that are born in a consanguineous family of Iranian descent.
As to clinical manifestations, it has been reported that cataract is the first symptom, which is frequently appears in the first decade while tendon xanthomas manifest in the second or third decade of life. However, in our patient, xanthomas were the initial manifestation on the spinal cord as well as seizures at the age of one. It should be mentioned that infantile spasms have been reported as an underappreciated symptom in infants with CTX [11].
As reported, they showed cognitive impairment from early infancy. Other highlighted signs included behavioral changes, dementia, hallucinations, aggression, agitation, and depression reported between puberty and the third decade of life. This has not appeared in our patients so far. In addition to the mentioned clinical symptoms, increased butyrylglycine and vomiting have been observed in our cases, which lead to misdiagnosis as a SCAD first. Vomiting is a rare clinical manifestation for CTX patients, which has been not reported previously. Furthermore, there is a lot of controversy about SCAD deficiency and, whether it should be considered a clinical entity or not is controversial. Many reports consider it a non-disease [12, 13]. Therefore, for the differential diagnosis, early genetic analysis in complex cases should be performed.
Early and correct diagnosis of CTX by next-generation sequencing (NGS) is crucial and effective. NGS is an accurate, efficient, and cost-effective method for the diagnosis of inherited metabolic disorders. It is documented that NGS can be useful for simultaneous sequencing of the group of candidate genes, especially for clinically and genetically heterogeneous diseases caused by a group of genes involving a common metabolic pathway. An early genetic diagnosis by NGS besides clinical suspicion, laboratory, biochemical confirmation of CTX, and imaging findings results in correct diagnosis and treatment, cost, and time effectiveness [14]. Until now, more than 250 variants are reported in CTX patients, and 85 variants are pathogenic or likely pathogenic [15]. Variants including missense, nonsense, frameshift, splice site, duplications, deletion, indel, and insertion have been reported so far. Variants were detected in all 9 exons and introns 2,4,6,7, and 8 of the CYP27A1 gene; however, the splice site variant had the highest prevalence, and exon 4 had the highest number of mutations, based on ClinVar database [16]. It is worth mentioning that the most prevalent variant among spinal CTX patients is reported to be the Arg395Cys allele [15].
It is important that when genetic analysis of CYP27A1 confirmed the CTX, its treatment by CDCA should be started as soon as possible. Treatment with CDCA has been documented to improve or even prevent clinical signs of CTX in the early stage of the disease. It can improve symptoms by direct inhibition of CYP7A1 hydroxylation of cholesterol and has a negative feedback on cholesterol biosynthesis, to decrease the rate of bile acid synthesis (and production of cholestanol) resulting in decreased accumulation of cholestenol in the tissues. It has appeared that early treatment by CDCA can prevent the development of neurologic symptoms in CTX patients [17].
In conclusion, our study reported two Iranian patients who suffered from CTX. A pathogenic variant in the CYP27A1 gene was identified, which is represented by a new clinical manifestation in our CTX patients. Regarding the progress and utility of NGS application, early detection before the onset and identifying more patients is predicted. It can also help to reduce the complications of this disease and increase our knowledge about rare inherited metabolic diseases.
Availability of data and materials
The data that support the findings of this study are available from Molecular Genetics Laboratory, but restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. Data are, however, available from the authors upon reasonable request and with permission of Molecular Genetics Laboratory. The datasets generated and/or analyzed during the current study are available in the Genbank repository (GRCh37/hg19, https://www.ncbi.nlm.nih.gov/genome/guide/human/) for CYP27A1: NG_007959.1.
Abbreviations
- CA:
-
Cholic acid
- CDCA:
-
Chenodeoxycholic acid
- CTX:
-
Cerebrotendinous xanthomatosis
- MR:
-
Mitral regurgitation
- NGS:
-
Next-generation sequencing
- SCAD:
-
Short-Chain acyl-CoA dehydrogenase
- WES:
-
Whole exome sequencing
References
Cali JJ, Hsieh CL, Francke U, Russell DW (1991) Mutations in the bile acid biosynthetic enzyme sterol 27-hydroxylase underlie cerebrotendinous xanthomatosis. J Biol Chem 266:7779–7783
Nie S, Chen G, Cao X, Zhang Y (2014) Cerebrotendinous xanthomatosis: a comprehensive review of pathogenesis, clinical manifestations, diagnosis, and management. Orphanet J Rare Dis 9:179
Lorincz MT, Rainier S, Thomas D, Fink JK (2005) Cerebrotendinous xanthomatosis: possible higher prevalence than previously recognized. Arch Neurol 62(9):1459–1463
Li ZR, Zhou YL, Jin Q, Xie YY, Meng HM (2022) CYP27A1 mutation in a case of cerebrotendinous xanthomatosis: a case report. World J Clin Cases 10(18):6168–6174
Baghbanian SM, MahdaviAmiri MR, Majidi H (2021) Cerebrotendinous xanthomatosis revisited. Pract Neurol 21:243–245
Mutlu D, Tuncer A, Gocmen R et al (2019) Diagnostic challenge: a case of late onset spinal form cerebrotendinous xanthomatosis. Neurology 92:438–439
Koyama S, Sekijima Y, Ogura M, Hori M, Matsuki K, Miida T, Harada-Shiba M (2021) Cerebrotendinous xanthomatosis: molecular pathogenesis, clinical spectrum, diagnosis, and disease-modifying treatments. J Atheroscler Thromb 28(9):905–925
Richards S, Aziz N, Bale S, Bick D (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med 17:405–423
Alhariri A, Hamilton K, Oza V, Cordoro K, Sobreira NL, Malloy M, Slavotinek A (2017) A patient with a late diagnosis of cerebrotendinous xanthomatosis and a response to treatment. Am J Med Genet 173(8):2275–2279
Chen SF, Tsai NW, Chang CC, Lu CH, Huang CR, Chuang YC, Chang WN (2011) Neuromuscular abnormality and autonomic dysfunction in patients with cerebrotendinous xanthomatosis. BMC Neurol 11:63
Larson A, Weisfeld-Adams JD, Benke TA, Bonnen PE (2017) Cerebrotendinous xanthomatosis presenting with infantile spasms and intellectual disability. JIMD Rep 35:1–5
Nochi Z, Olsen R, Gregersen N (2017) Short-chain acyl-CoA dehydrogenase deficiency: from gene to cell pathology and possible disease mechanisms. J Inherit Metab Dis 40(5):641–655
van Maldegem BT, Duran M, Wanders R, Niezen-Koning K, Hogeveen M, Ijlst L et al (2006) Clinical, biochemical, and genetic heterogeneity in short-chain acyl-coenzyme A dehydrogenase deficiency. JAMA 296(8):943–952
Beyzaei Z, Geramizadeh B, Karimzadeh S (2020) Diagnosis of hepatic Glycogen storage disease patients with overlapping clinical symptoms by massively parallel sequencing: a systematic review of literature. Orphanet J Rare Dis 15:217
Atallah I, Millán DS, Benoît W, Campos-Xavier B, Superti-Furga A, Tran C (2021) Spinal cerebrotendinous xanthomatosis: a case report and literature review. Mol Genet Metab Rep 26:100719
Appadurai V, DeBarber A, Chiang PW, Patel SB, Steiner RD, Tyler C, Bonnen PE (2015) Apparent underdiagnosis of Cerebrotendinous Xanthomatosis revealed by analysis of ~60,000 human exomes. Mol Genet Metab 116(4):298–304
Stelten BML, Huidekoper HH, van de Warrenburg BPC, Brilstra EH, Hollak CEM, Haak HR, Kluijtmans LAJ, Wevers RA, Verrips A (2019) Long-term treatment effect in cerebrotendinous xanthomatosis depends on age at treatment start. Neurology 92(2):e83–e95
Acknowledgments
The authors hereby extend their gratitude to the patients for their participation in the present study. We appreciate the data base registry of Metabolic (no. 1400-7860), Shiraz University of Medical Sciences.
Funding
Nothing to declare.
Author information
Authors and Affiliations
Contributions
ZB contributed to conception of the manuscript, acquisition of data, drafting and final approval the manuscript. HM contributed to acquisition of data, revising the manuscript. SI contributed to revising the manuscript. MHI contributed to acquisition of data, revising the manuscript. BG contributed to acquisition of data, revision and final approval of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was conducted in accordance with the Declaration of Helsinki. It was approved by the Ethics Committee of Shiraz University of Medical Sciences. The researcher informed the parents of the children about the objectives of the study, examination and investigations that have been done. Also, the confidentiality of their information, their rights not to participate in the study were respected and written informed consent was obtained.
Consent for publication
Written informed consent was obtained from their parent or legal guardian for publication of this case report and accompanying images.
Competing interests
The authors declare that they have no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Beyzaei, Z., Moravej, H., Imanieh, M.H. et al. Identification of two Iranian siblings with cerebrotendinous xanthomatosis: a case report. Egypt J Med Hum Genet 24, 34 (2023). https://doi.org/10.1186/s43042-023-00413-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-023-00413-1