
Abstract
Background
Charcot–Marie–Tooth disease comprises a large spectrum of clinically heterogeneous disorders. PLEKHG5 variants have shown an intermediate form of autosomal-recessive Charcot–Marie–Tooth disease C and distal spinal muscular atrophy IV. The purpose of this case study is to report a causative genetic defect associated with intermediate form of autosomal-recessive Charcot–Marie–Tooth disease C in an Iraqi consanguineous family.
Case presentation
Whole-exome and Sanger sequencing was used to identify probable gene defects in a 9-year-old male affected by CMTRIC. We found a new single mutation (c.1844C > A; p.T615N) in the PLEKHG5 gene, located in exon 17 (NM_020631.6), causing a missense mutation that has been changed one amino acid. The mutation was homozygous in the patient and heterozygous in his parents.
Conclusion
Our results expand the PLEKHG5 pathogenic mutation spectrum related to intermediate form of autosomal-recessive Charcot–Marie–Tooth disease C which is vital for screening and genetic diagnosis of the disease.
Similar content being viewed by others

Background
Charcot–Marie–Tooth (CMT) disease, also known as hereditary motor and sensory neuropathy (HMSN), encompasses a genetically and clinically heterogeneous group of disorders characterized by weakness, muscular wasting, and sensory loss usually most severe distally. At least 1 in 2,500 people are affected by this inherited neuromuscular disorder [1].
There are now > 100 genetic mutations identified as causative for CMT. Based on neurophysiology and nerve biopsy, CMT is classified into two subtypes, demyelinating forms (CMT1) and axonal forms (CMT2). An intermediate group also exists with nerve conduction velocities, which overlaps the two main groups, and is subdivided into recessive intermediate CMT (RI-CMT) and dominant intermediate (DI-CMT) by its pattern of inheritance [2].
Pleckstrin homology domain-containing, family G member 5 (PLEKHG5) gene encodes a guanine exchange factor specifically expressed in motor neurons, where it regulates the autophagy of synaptic vesicles. PLEKHG5 gene plays essential role in the activation and regulation of RhoA signaling pathways, thereby controlling neuronal cell differentiation and maintaining stable cell–cell contacts. Moreover, mutations in the PLEKHG5 gene are causing different forms of motor neuron diseases such as distal spinal muscular atrophy IV (DSMA4, also known as lower motor neuron diseases [LMND]) and intermediate Charcot–Marie–Tooth disease type C (CMTRIC), both of which inherited in an autosomal-recessive pattern [3, 4]. Autosomal-recessive CMTRIC is a very rarely seen neurogenetic disorder [5]. Whole-exome sequencing (WES) technique is a particularly useful tool for understanding the molecular genetic causes of the CMT [6]. According to this evidence, we employed WES to identify the likely disease-causing genetic variants in a consanguineous Iraqi family affected by CMT. Here, we present the first consanguineous CMTRIC case due to a PLEKHG5 substitution mutation not previously described.
Case presentation
In the present study, there is an Iraqi family with a 9-year-old male patient (Fig. 1), who was referred to the pediatric neurologists due to frequent outdoor falls and difficulty in climbing up stairs. The proband was determined to have a clinical diagnosis consistent with CMT. His parents mentioned that he had normal motor and mental development compared to his peers during infancy and childhood. In addition, the patient had not a family history of neuromuscular diseases. Neurologic examination showed limb–girdle muscular weakness. His parents also noted that he started experiencing difficulty in getting up from squatting position. Physical examination revealed scapula winging and absent deep reflexes. Creatine kinase (CPK) levels were slightly elevated, and metabolic screening of urine, lactate, and pyruvate levels was all within normal limits. During follow-up, motor regression was noted with a positive Gowers’ sign with a longer duration. In addition, he could not stand up from a sitting position without support. Neurological examination revealed atrophy of bilateral distal muscles and muscle weakness, predominantly in the lower limbs. He had a 25-degree thoracolumbar scoliosis. Ophthalmic examination and hearing tests were in normal ranges, and also, his cardiac evaluation, including an echocardiogram and electrocardiogram, was within normal limits.

Pedigree of family. Patient 1 was a 9-year-old affected male in the presented study. The parents of the affected son (3,4) are first cousins
At his electrophysiological examination, motor nerve conduction studies (NCSs) and F-wave studies were performed for the right tibial, ulnar, and peroneal nerves, and sensory NCSs were performed for the right median, ulnar, and bilateral sural nerves using standard techniques. F-wave latencies were significantly prolonged in the median and tibial motor nerves. The ulnar and median motor and sensory nerve conduction velocities were slow. The ulnar and median motor and ulnar sensory nerve conduction velocities were within normal ranges, while the median sensory nerve conduction velocity was slow. Bilateral sural nerve sensory action potentials could be obtained, but with very low amplitudes. Fibrillation potentials and positive sharp waves were both significant active and chronic neurogenic findings for the tibialis anterior, biceps, iliopsoas, and first dorsal interosseous muscles with needle electromyography (EMG) examination. Myokymic discharges were found in the biceps muscle of the proband. The electrophysiological study suggested a predominantly axonal sensory and motor polyneuropathy and some demyelinating features. Finally, he was diagnosed as having CMT and we applied WES technique to identify the likely disease-causing mutations in the patient’s DNA that may be inherited from the patient’s ancestors.
We solely performed WES for the proband. For genomic DNA (gDNA) extraction from peripheral blood, a standard salting out method was used. The patient’s DNA was captured using SureSelect Human All Exome Kit V6 (Agilent Technologies Inc., Santa Clara, CA, USA) and sequenced those regions using the Illumina HiSeq 4000 machine (San Diego, CA, USA) as stated by the manufacturer’s protocols. The average read depth was more than 100 ×, and depth of 20 × or greater was achieved in 98.0% of the targeted genomic sequence. We detected a novel missense substitution mutation in exon 17 of PLEKHG5 gene (NM_020631.6: c.1844C > A; p.Thr615Asn), which indicates that this mutation is associated with CMTRIC. This mutation predicts an alteration in codon translation, and finally a change in one amino acid (threonine converted to asparagine).
Furthermore, the c.1844C > A; p.Thr615Asn mutation was classified as likely pathogenic according to the American College of Medical Genetics and Genomics (ACMG) guidelines. Bioinformatics software such as MutationTaster, PolyPhen-2, SIFT, and CADD predicted that this mutation will be deleterious and disease-causing.
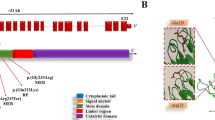
To co-segregate and confirm the presence of the pathogenic PLEKHG5 mutation, direct Sanger sequencing method for the patient and his parents was done. So, we have designed a specific set of primer pairs (forward primer: GGCAGCCATATTCCACAAGT and the reverse primer: GGGCATTGTAAATGGTGTCC) to amplify the mutated site in the genome by PCR method. After amplification of PLEKHG5 sequences, we sequenced the PCR products directly on the automated genetic analyzer (Applied Biosystems 3130xl; USA) and the results are represented in Fig. 2. The sequences were blasted in NCBI website (http://www.ncbi.nlm.nih.gov/blast) and compared with normal sequences. Interestingly, this finding has not been reported in the other patients. Figure 2D shows the location of the novel variant of PLEKHG5 protein. The mutant amino acids are located in the highly conserved PH domain of PLEKHG5 gene, which includes a domain in 584–684 [3].

Sanger sequencing confirming homozygous c.1844C > A mutation in the patient (A) and heterozygous mutation in his parents (B, C). D Diagram structure of PLEKHG5 protein. Schematic of PLEKHG5 protein shows the locations of the pathogenic variant in humans
Discussion
In this study, WES method was applied to detect the causative genes defects related to CMT in an Iraqi family. The index patient was a homozygous carrier for PLEKHG5 T615N mutation, and the parents of the patient were both heterozygous. Therefore, the NM_000249 (PLEKHG5): c.1844C > A mutation is surely responsible for CMTRIC phenotype in the proband. This is the first Iraqi case of a CMTRIC phenotype caused by PLEKHG5 gene mutation. So far, eight different PLEKHG5 mutations have been recognized based on the Human Gene Mutation Database (HGMD; http://www.hgmd.cf.ac.uk/ac/index.php), one of which presented childhood-onset LMND, whereas the other seven presented intermediate CMT, amyotrophic lateral sclerosis (ALS), and myopathy.
Hence, in this study, we investigated the disease-causing mutation in a 9-year-old affected male, who had limb–girdle muscle weakness, and this condition was reported in almost all cases [7]. Distal sensory loss was reported in almost 1/3 of the patients. All of the patients had distal muscle weakness in the lower limbs. In contrast, in the current study, the patient had proximal muscle weakness in the lower extremities. However, the presence of areflexia, distal muscle atrophy, spinal cord abnormalities, and foot deformity was consistent with other defined cases.
Electrophysiological studies of patients showed prolonged motor latencies and their median motor nerve conduction velocities were slowed, the amplitudes of the sensory nerve action potentials (SNAP) were absent or decreased, and their conduction velocities were sufficiently slowed in the affected nerves. Needle EMG showed muscle denervation in almost all of the patients [8, 9]. In the present case, intermediate polyneuropathy was detected moderately with predominantly axonal features evident with active and chronic denervation findings.
PLEKHG5 is predominantly expressed in the human peripheral nervous system, and the protein contains a Dbl homology (DH)–Pleckstrin homology (PH) motif, which is known as the minimal unit required for the nucleotide-exchange-promoting function of guanine nucleotide-exchange factors (GEFs) [2]. Recessive mutations in the PH domain and PLEKHG5 gene were initially reported in a consanguineous family with early-onset diseases of the lower motor neuron (or DSMA4) and formally have been associated with intermediate CMT. In more studies, cases with either pure LMND, distal and proximal neuropathy with mild sensory involvement, or intermediate CMT have been documented [10]. Additionally, we reported the case of a 9-year-old Iraqi male who presented with CMTRIC with a novel homozygous mutation in PLEKHG5 gene (c.1844C > A; p.Thr615Asn), and this detected mutation affects a conserved residue and supports a positive pathogenic role for this gene in causing the CMTRIC.
PLEKHG5’s molecular functions and disease-associated mechanisms are limited: Previously, only one homozygous substitution mutation, c.1940 T > C; p.Phe647Ser, was reported as a pathogenic mutation in PLEKHG5 gene associated with autosomal-recessive LMND. The site locates within the PH domain, and the mutation was suggested to affect the nuclear factor kappa B (NF-κB) activation. According to immunohistochemical results from the patient and immunocytochemical results obtained from mutants that have been cloned, mutant proteins have lower expression levels than wild-type proteins. As a result, these data suggest that the patient with peripheral neuropathy is due to low levels of expression and NF-κB activation [2, 11]. Also, we proposed that our identified Thr615Asn variant encodes an impaired PLEKHG5 protein that probably causes a defect in the activation of the NF-κB signaling pathway resulting in the CMTRIC.
Some studies revealed that mutations in PLEKHG5 including c.1600-2A > G, c.1835_1860del, c.2308del and c.104del are associated with intermediate CMT disease [5, 10]. In this regard, Beijer D. et al. identified a homozygous single PLEKHG5 mutation (p.Arg97Gln), predicting an alteration in codon translation (arginine converted to glutamine), which affects the PLEKHG5 protein stability, resulting in CMT clinical manifestations [12]. Moreover, Kim HJ. et al., in their publication, reported a case of autosomal-recessive intermediate CMT disease with novel compound heterozygous (p.Thr663Met and p.Gly820Arg) variants in the PLEKHG5 gene [2]. Our report adds a novel variant to PLEKHG5 mutation spectrum and indicates that a missense c.1844C > A; p.Thr615Asn mutation probably causes PLEKHG5 dysfunction leading to the CMTRIC.
Finally, this detected mutation that inherited from patient’s ancestors is proposed as the cause of CMTRIC in the patient and the following evidence proves that this mutation can lead to the disease: 1—WES identified only this mutation as the cause of CMTRIC. 2—As can be seen in Fig. 2A, direct Sanger sequencing proved the mutation in the proband, and based on recognized heterozygote mutations in his parents, the pattern of inheritance must be an autosomal-recessive for the PLEKHG5 gene. 3—Bioinformatics software such as MutationTaster, PolyPhen-2, SIFT, and CADD predicted that c.1844C > A mutation is the pathogenic mutation for CMTRIC. 4—Also, a substitution mutation in exon 17 of the PLEKHG5 gene (c.1844C > A; p.Thr615Asn) in the PH domain of the protein (Fig. 2D), which is predicted to create new codon substitutions, can create a major problem in the PLEKHG5 protein. Based on our results, WES is an efficient approach of analyzing a patient’s DNA to discover the genetic cause of CMTRIC.
Conclusion
We have successfully applied WES in an Iraqi family for mutation screening within CMT-related genes and identified a novel substitution mutation in the PLEKHG5 gene, thereby confirming previous reports that PLEKHG5 gene mutations are associated with CMTRIC. This study may be helpful in providing appropriate genetic counseling to the affected families and provides a new way to clarify the molecular mechanisms of the PLEKHG5 gene in CMTRIC.
Availability of data and materials
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Abbreviations
- ACMG:
-
American College of Medical Genetics and Genomics
- CMTRIC:
-
Intermediate Charcot–Marie–Tooth disease type C
- CPK:
-
Creatine kinase
- DH:
-
Dbl homology
- DSMA4:
-
Distal spinal muscular atrophy IV
- EMG:
-
Electromyography
- gDNA:
-
Genomic DNA
- GEFs:
-
Guanine nucleotide-exchange factors
- HMSN:
-
Hereditary motor and sensory neuropathy
- LMNDs:
-
Lower motor neuron diseases
- NF-kB:
-
Nuclear factor kappa B
- NCSs:
-
Nerve conduction studies
- PH:
-
Pleckstrin homology
- SNAP:
-
Sensory nerve action potentials
- WES:
-
Whole-exome sequencing
References
Reilly MM, Murphy SM, Laurá M (2011) Charcot-Marie-Tooth disease. J Peripher Nerv Syst 16(1):1–14. https://doi.org/10.1111/j.1529-8027.2011.00324.x
Kim HJ, Hong YB, Park JM, Choi YR, Kim YJ, Yoon BR, Koo H, Yoo JH, Kim SB, Park M, Chung KW, Choi BO (2013) Mutations in the PLEKHG5 gene is relevant with autosomal recessive intermediate Charcot-Marie-Tooth disease. Orphanet J Rare Dis 12(8):104. https://doi.org/10.1186/1750-1172-8-104
Gonzalez-Quereda L, Pagola I, Fuentes Prior P, Bernal S, Rodriguez MJ, Torné L, Salgado Garrido J, Gallano P, Jericó I (2021) Novel PLEKHG5 mutations in a patient with childhood-onset lower motor neuron disease. Ann Clin Transl Neurol 8(1):294–299. https://doi.org/10.1002/acn3.51265
Miao Y, Yu M, Meng L, Zhang W, Lv H, Wang Z, Yuan Y (2021) PLEKHG5-related autosomal recessive lower motor neuron disease with dysmyelination in peripheral nerves. Clin Neuropathol 40(6):328–332. https://doi.org/10.5414/NP301377
Yayici Köken Ö, Öztoprak Ü, Topçu V, Çavdarli B, Temuçin ÇM, Aydingöz Ü et al (2021) Expanding the genotype-phenotype spectrum of autosomal recessive charcot-marie-tooth disease: a novel plekhg5 gene mutation. Neurol Asia 26(3):607–612
Choi BO, Koo SK, Park MH, Rhee H, Yang SJ, Choi KG, Jung SC, Kim HS, Hyun YS, Nakhro K, Lee HJ, Woo HM, Chung KW (2012) Exome sequencing is an efficient tool for genetic screening of Charcot-Marie-Tooth disease. Hum Mutat 33(11):1610–1615. https://doi.org/10.1002/humu.22143
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, ACMG Laboratory Quality Assurance Committee (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–424. https://doi.org/10.1038/gim.2015.30
Chen Z, Maroofian R, Başak AN, Shingavi L, Karakaya M, Efthymiou S, Gustavsson EK, Meier L, Polavarapu K, Vengalil S, Preethish-Kumar V, Nandeesh BN, Gökçe Güneş N, Akan O, Candan F, Schrank B, Zuchner S, Murphy D, Kapoor M, Ryten M, Wirth B, Reilly MM, Nalini A, Houlden H, Sarraf P (2021) Novel variants broaden the phenotypic spectrum of PLEKHG5-associated neuropathies. Eur J Neurol 28(4):1344–1355. https://doi.org/10.1111/ene.14649
Saporta AS, Sottile SL, Miller LJ, Feely SM, Siskind CE, Shy ME (2011) Charcot-Marie-Tooth disease subtypes and genetic testing strategies. Ann Neurol 69(1):22–33. https://doi.org/10.1002/ana.22166
Villar-Quiles RN, Le VT, Leonard-Louis S, Trang NT, Huong NT, Laddada L, Francou B, Maisonobe T, Azzedine H, Stojkovic T (2021) Leukoencephalopathy and conduction blocks in PLEKHG5-associated intermediate CMT disease. Neuromuscul Disord 31(8):756–764. https://doi.org/10.1016/j.nmd.2021.06.004
Maystadt I, Rezsöhazy R, Barkats M, Duque S, Vannuffel P, Remacle S, Lambert B, Najimi M, Sokal E, Munnich A, Viollet L, Verellen-Dumoulin C (2007) The nuclear factor kappaB-activator gene PLEKHG5 is mutated in a form of autosomal recessive lower motor neuron disease with childhood onset. Am J Hum Genet 81(1):67–76. https://doi.org/10.1086/518900
Beijer D, Polavarapu K, Preethish-Kumar V, Bardhan M, Dohrn MF, Rebelo A, Züchner S, Nalini A (2022) Homozygous N-terminal missense variant in PLEKHG5 associated with intermediate CMT: a case report. J Neuromuscul Dis 9(2):347–351. https://doi.org/10.3233/JND-210716
Acknowledgements
The authors would like to thank the family members for their participation in this study.
Funding
None.
Author information
Authors and Affiliations
Contributions
MN, HM, AIA, JMA, and RAA analyzed and interpreted the data. MN wrote the manuscript. HM edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All procedures performed in this study were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or compare ethical strand.
Consent for publication
Written informed consent was obtained from the family for this publication.
Competing interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Neissi, M., Mabudi, H., Al-Badran, A.I. et al. A novel missense mutation in PLEKHG5 gene causing an intermediate form of autosomal-recessive Charcot–Marie–Tooth disease in an Iraqi family. Egypt J Med Hum Genet 24, 25 (2023). https://doi.org/10.1186/s43042-023-00403-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-023-00403-3