
Abstract
Background
Intellectual developmental disorder with dysmorphic facies and ptosis (MIM #617333) is a very rare condition, characterized by more than 80% by language delay, intellectual disability, gross motor development delay, broad nasal bridge, hypertelorism, and hypotonia. This condition exhibits as autosomal dominant inheritance and is caused by a heterozygous variant in the BRPF1 gene. Additionally, the copy number variation in the terminal region of chromosome 3p (MIM #613792) has been shown to manifest in most patients as intellectual disability, motor delay, and hypotonia.
Case presentation
We present an 18-year-old male patient with facial dysmorphism, intellectual disability, ptosis, and congenital heart disease. Using chromosomal microarray analysis, a previously unreported 90 kb deletion involving seven genes was found.
Conclusion
When comparing our findings with 39 previous reports, we found that the common clinical features of this syndrome, such as gross motor delay, hypotonia, and congenital spinal cord abnormalities, were not observed in this patient. From the seven genes implicated in the deletion, only BRPF1 could be strongly correlated with the phenotype, according to its function and haploinsufficiency coefficients.
Similar content being viewed by others

Background
Intellectual disability (ID) manifests before the age 18 years and is defined as a limitation in two areas: intelligence or mental capability, and adaptive behavior in any of its three domains (conceptual, social, and practical) [1]. The global prevalence of ID is 1–3%, and the copy number variations (CNV) are the cause of ID in 36.1% of these cases [2]. To date, more than 2000 genes associated with ID have been reported (https://www.sysid.dbmr.unibe.ch). Of these, BRPF1 gene (Bromodomain and PHD finger -containing 1) has been previously associated with an intellectual developmental disorder with dysmorphic facies and ptosis, or IDDDFP (MIM #617333), which is characterized by neonatal feeding disorder, hypotonia, gross and fine motor development delay, language delay, intellectual disability, epilepsy, brain abnormalities, flat and round face, broad nasal bridge, hypertelorism, small palpebral fissures, ptosis, blepharophimosis, joint hypermobility, and spinal anomalies [3]. To date, 39 patients with IDDDFP have been reported, most of them diagnosed in the USA and Europe with variants in the BRPF1 gene detected by exome sequencing [3,4,5,6,7,8,9,10].
The BRPF1 (3p26-p25) has 14 exons and transcribes four isoforms. This gene code for a protein is called peregrin, which contains 1214 amino acids [11] and is a scaffolding subunit of several histone acetyltransferases, such as MOZ (KAT6A)/MORF(KAT6B) and the HBO1(KAT7) complex, having an acetyltransferase activity in the H3 histone [3, 12, 13]. Previous research suggests that MOZ/MORF, or HBO1 together with peregrin, EAF6, and ING5, bind transcription factors like Runx and p53, and intervenes in the embryonic development of hematopoietic and neuronal cells. Furthermore, variants in these molecules have also been related to leukemia, malignant neoplasms, and ID [14].
Here, we present the first Peruvian and Latin-American patient with a suspected case of IDDDFP caused by a CNV present in chromosome 3. The deletion includes the BRPF1 gene, which adds evidence to the studies in animal models which show it is a haploinsufficient gene [15]. In addition, we highlight the importance of applying genome techniques, like CMA, to diagnose neurodevelopmental disorders in low- and middle-income countries.
Case presentation
Clinical report
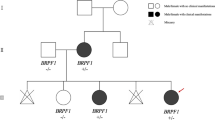
The patient is an 18-year-old male with non-consanguineous parents, without a family history of neurodevelopment disorders, and with an uneventful prenatal history. His mother presented with preeclampsia during pregnancy, causing the delivery to be carried out by cesarean section at 38 weeks of gestation, with a birth weight of 2.7 kg and an apparently normal Apgar score. However, the patient’s height and head circumference were not recorded. Psychomotor development showed that he achieved head control at two months, sitting without support at six months, and walking at one year of age. Regarding his speech development, he said his first words at one year of age, and he began to form sentences at five years old, with a social smile at two months. The patient has a history of atrial septal defect (ASD) and a diagnosis of acquired hypothyroidism at ten years of age (under treatment with levothyroxine at 50 μg/day) with normal values of TSH and free-T4 at the last control. He underwent surgery for ASD twice (at 12 and 14 years of age) and tympanic membrane perforation (at 15 and 16 years of age). Additionally, he presented hyperglycemia and acanthosis nigricans, for which he is still medicated with metformin. In childhood, he was diagnosed with hyperactivity, and he reached eleventh grade with low performance. However, there is no family history of neurodevelopmental disorders or congenital anomalies (Fig. 1A).

Source: Own elaboration
A Pedigree indicated a de novo variant (III.7) in this family, paternal and maternal ancestry are from different regions B Palpebral ptosis, bilateral epicanthus, blepharophimosis, C Prominent columella, short philtrum, thick vermilion of the lips. D Brachydactyly and decrease in the distal interphalangeal crease of the fourth finger. E Normal function of BRPF1 gene (peregrin). F Haploinsufficiency of BRPF1 produces a reduction of various processes in the brain. BRPF1 = Bromodomain- and PHD finger- containing protein. ING5 = Inhibitor of Growth 5. MEAF6 = Myst/Esa1-associated factor 6. KAT6A/B = Lysine acetyltransferase 6A and 6B. KAT7 (HBO1) = Lysine acetyltransferase 7. H3 = histone 3.
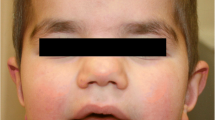
Physical examination showed that the patient presented normal anthropometry, narrow forehead, blepharophimosis, palpebral ptosis, short and deep philtrum, thick vermilion of the lips, underdeveloped supraorbital ridges, brachydactyly, and limitation of the flexion of the fourth finger (Fig. 1B–D). Among the complementary evaluations, he had an IQ of 63.
Chromosomal microarray analysis (CMA)
For the molecular analysis and the publication of this article, informed consent of the parents was requested. According to the manufacturer, the chromosomal microarray analysis was performed using genomic DNA, which was amplified, labeled, and hybridized based on the GeneChip CytoScan 750 K Array (Affymetrix, USA ®) instructions. The DNA genomic was extracted from a blood sample using the kit gSYNC™ DNA Extraction, following the manufacturer’s instructions. DNA concentration and purity were measured with spectrophotometer Nanodrop 2000 (Thermo Scientific, USA). Briefly, 250 ng of DNA was digested with restriction enzyme Nsp I and was then linked with adapters for the PCR amplification. The products of the PCR were analyzed with electrophoresis with agarose gel 2% E-Gel ® EX (Invitrogen, USA), and were posteriorly purified using magnetic beads. DNA purified was fragmented using DNAsa 1 and analyzed trough electrophoresis with agarose gel 4% E-Gel®EX (Invitrogen, USA). Fragmented products were labeled with biotin and hybridized for 18 h at the genechip; next, these were washed and colored with fluid station Affymetrix 450. The test included 550,000 non-polymorphic markers and 200,436 single nucleotide polymorphisms (SNP) markers. Finally, the gene chips were scanned with Affymetrix 3000 GeneChip Scanner and analyzed with Chromosome Analysis Suite (ChAS) v.4.2 (Affymetrix, USA®). The gains or losses were considered for analysis when at least 50/25 markers were compromised, respectively. In addition, the regions of homozygosity (ROH) were considered for analysis when the length was at least 5 Mb (ver Thermo Fisher Sc Inc, 2017).
The CMA result for the patient was arr[GRCh38] 3p25.3(9706732_9796589) × 1, with a ROH percentage of 0,69. The CNV, a 90 kb deletion, contained seven genes: CPNE9, BRPF1, OGG1, CAMK1, TADA3, ARPC4, and ARPC4-TTLL3. We did not perform another genomic test (i.e. whole exome sequencing) because CMA showed a variant related to the phenotype.
Discussion
Haploinsufficiency is the mechanism by which a gene in a hemizygous state causes a phenotype [16]. A likely cause of haploinsufficiency is that some genes that present conserved sequences during evolution with few functional coding variants are more likely to have a dose-sensitive effect [17]. The monoallelic expression occurs only when one allele needs to be expressed for a given function, and around 3000 genes have been established in humans. Conversely, in the biallelic expression, it is necessary that both alleles of a gene function simultaneously, and therefore the latter have a lower tolerance to loss in CNVs [18]. It has also been observed that in the process of evolution, during the complete genome duplication, some genes were retained, and others lost; the retained genes are called ohnologues, which are known to be involved in the embryonic development and are part of protein complexes [19].
A CMA performed on the patient showed the deletion of a chromosome fragment that contained seven genes were present in a single copy and probably de novo. All these genes require both alleles to be present to function correctly. Of these, only BRPF1 has been associated with a genetic disease (Additional file 1). This gene codes for peregrin, a multivalent protein chromatin reader that interacts with the histone acetyltransferase and activates them (epigenetic regulation), acting in a complex to promote the acetylation of lysine 23 of histone 3 [3]. Studies in mice and zebrafish indicate that BRPF1 is essential for the embryo’s survival, hematopoiesis, head pattern, and brain development. It is expressed in the neocortex, hippocampus, cerebellum, and olfactory bulb [20,21,22].
BRPF1 has a haploinsufficiency coefficient of 15.77%, and with a loss of function observed/expected upper bound fraction coefficient (LOEUF) of 0.176 [23, 24]. In mice, it has been observed that BRPF1 haploinsufficiency causes reduction of the dendritic complex in hippocampal granulosa cells and the cortical pyramidal neurons, as well as reduction in the density and morphology of the dendritic column (Fig. 1E, F) [15].
To date, the other genes included in the CNV carried by the patient described in this paper (Additional file 1) have not been associated with a disease. For example, the OGG1 gene (8-oxoguanine DNA glycosilase) encodes an enzyme related to transcriptional regulation and the maintenance of metabolic homeostasis, and heterozygous somatic variants have been associated with renal cell carcinoma (MIM #144700) [25, 26]. Two of the other implicated genes could nevertheless be eventually associated with the patient’s phenotype: Cpen9 and ARPC4. Cpen9 (copine family member 9) has been related to calcium turnover, and it can be associated with cognitive performance. However, its probability of being loss-of-function intolerant (pLI) is very low (0.001), and the LOEUF is greater than 0.35 [27]. In this sense, the ARPC4 has a pLI of 0.938 and a LOEUF of 0.265, and its function is to mediate actin polymerization through the stimulation by promoter factor nucleation and inhibition expression, which significantly attenuates the proliferation, migration, and the invasion in bladder cancer [28, 29].
Therefore, we believe that a considerable part of the described patient’s phenotype is related to the intellectual development disorder with dysmorphic facies and ptosis, or IDDDFP (MIM # 617333) [7, 30]. It is characterized by microcephaly, short stature, brain abnormalities, seizures, strabismus, joint hypermobility, fusion of cervical vertebral bodies, camptodactyly, and short metacarpal, among other symptoms [7]. To date, 39 IDDDFP patients with 18 basic clinical characteristics have been described. The most common pathogenic variants in the BRPF1 gene were substitution, followed by deletion and intragenic duplication; however, no variants with complete deletion of the gene have so far been registered (Table 1). The patient described in this study presents ten of the 18 clinical characteristics mentioned above. These include palpebral ptosis and blepharophimosis which was the main leading clinical features to suspect the presence of this disorder; however, according to the reported cases, blepharophimosis and ptosis are observed in 63.6% and 56.8% cases, respectively, while other nonspecific characteristics, such as language delay or intellectual disability, are the most frequently described characteristics. Therefore, it is difficult to establish a gestalt phenotype based on the cases reported so far. This highlights the importance of genomic diagnosis tools that allow the description of pathogenic variants.
From the CNV perspective, 31 patients with interstitial and terminal deletions in chromosome 3p have been previously described with phenotypic characteristics like those presented by this patient. The characteristics of the 3p deletion syndrome (MIM #613792) are ID, motor delay, microcephaly, micrognathia, ptosis, long philtrum, polydactyly, hypotonia; heart, renal, and gastrointestinal anomalies; hypothyroidism, epilepsy, short stature, and risk of tumors [7, 31,32,33]. However, as it is a contiguous gene syndrome, the clinical characteristics will be variable and depend on the number of genes involved. Out of the ten most important characteristics of the 3p deletion syndrome, our patient has only four (Additional file 1): ID, broad nose, palpebral ptosis, and heart anomalies.
Considering all the available information and results, we believe that our patient’s diagnosis is more similar to IDDDFP de novo than the 3p deletion syndrome, primarily because the CNV found involves only one gene that could be related to the clinical manifestations, either by function, its pLI, or the haploinsufficiency coefficient. Nevertheless, studies on issues such as RNA or proteins expression of the other six genes contained in the deletion (CPNE9, OGG1, CAMK1, TADA3, ARPC4, and ARPC4-TTLL3) should be carried out to know specifically how haploinsufficiency in these genes affect the phenotype of the patient.
Although parental consanguinity was not declared and the maternal and paternal grandparents come from different regions of Peru, the ROH was 0.69%, corresponding to a parental consanguinity relationship of the fifth degree.
Conclusions
To summarize, we suggest that dysmorphic features such as ptosis or blepharophimosis, if present from early years, could be considered as sufficient signs for the search for DNA pathogenic variants. Furthermore, our approach shows how imperative it is to use the molecular diagnosis in patients with these clinical features and how necessary it is to make these technologies more accessible. These tests can assist for determine of both the etiology and the prognosis, as well as the risk of recurrence.
Availability of data and materials
Data such as chromosomal microarray analysis or medical history supporting the findings of this study are available and may be obtained from the corresponding author upon reasonable request.
Abbreviations
- CMA:
-
Chromosomal microarray analysis
- IDDDFP:
-
Intellectual developmental disorder with dysmorphic facies and ptosis
References
Boat TF, Wu JT. Disorders C to E the SSIDP for C with M, Populations B on the H of S, Board on Children Y, Medicine I of, et al. Clinical characteristics of intellectual disabilities. US: National Academies Press; 2015.
Abarca-Barriga H, de Sotomayor MV, Trubnykova M, Velasquez FC, Pastor MAC, Jugo BEG et al (2020) Variantes en el número de copias en trastornos del neurodesarrollo, síndrome malformativo y talla baja en Perú. Acta Med Peru. https://doi.org/10.35663/amp.2020.372.915
Yan K, Rousseau J, Littlejohn RO, Kiss C, Lehman A, Rosenfeld JA et al (2017) Mutations in the chromatin regulator gene BRPF1 cause syndromic intellectual disability and deficient histone acetylation. Am J Hum Genet 100:91–104. https://doi.org/10.1016/j.ajhg.2016.11.011
Baker SW, Murrell JR, Nesbitt AI, Pechter KB, Balciuniene J, Zhao X et al (2019) Automated clinical exome reanalysis reveals novel diagnoses. J Mol Diagn JMD 21:38–48. https://doi.org/10.1016/j.jmoldx.2018.07.008
Demeulenaere S, Beysen D, De Veuster I, Reyniers E, Kooy F, Meuwissen M (2019) Novel BRPF1 mutation in a boy with intellectual disability, coloboma, facial nerve palsy and hypoplasia of the corpus callosum. Eur J Med Genet 62:103691. https://doi.org/10.1016/j.ejmg.2019.103691
Keywan C, Holm IA, Poduri A, Brownstein CA, Alexandrescu S, Chen J et al (2020) A de novo BRPF1 variant in a case of sudden unexplained death in childhood. Eur J Med Genet 63:104002. https://doi.org/10.1016/j.ejmg.2020.104002
Mattioli F, Schaefer E, Magee A, Mark P, Mancini GM, Dieterich K et al (2017) Mutations in histone acetylase modifier BRPF1 cause an autosomal-dominant form of intellectual disability with associated ptosis. Am J Hum Genet 100:105–116. https://doi.org/10.1016/j.ajhg.2016.11.010
Naseer MI, Abdulkareem AA, Guzmán-Vega FJ, Arold ST, Pushparaj PN, Chaudhary AG et al (2020) Novel missense variant in heterozygous state in the BRPF1 gene leading to intellectual developmental disorder with dysmorphic facies and ptosis. Front Genet 11:368. https://doi.org/10.3389/fgene.2020.00368
Pode-Shakked N, Barel O, Pode-Shakked B, Eliyahu A, Singer A, Nayshool O et al (2019) BRPF1-associated intellectual disability, ptosis, and facial dysmorphism in a multiplex family. Mol Genet Genomic Med 7:e665. https://doi.org/10.1002/mgg3.665
Yan K, Rousseau J, Machol K, Cross LA, Agre KE, Gibson CF et al (2020) Deficient histone H3 propionylation by BRPF1-KAT6 complexes in neurodevelopmental disorders and cancer. Sci Adv. https://doi.org/10.1126/sciadv.aax0021
Thompson KA, Wang B, Argraves WS, Giancotti FG, Schranck DP, Ruoslahti E (1994) BR140, a novel zinc-finger protein with homology to the TAF250 subunit of TFIID. Biochem Biophys Res Commun 198:1143–1152. https://doi.org/10.1006/bbrc.1994.1162
Doyon Y, Cayrou C, Ullah M, Landry A-J, Côté V, Selleck W et al (2006) ING tumor suppressor proteins are critical regulators of chromatin acetylation required for genome expression and perpetuation. Mol Cell 21:51–64. https://doi.org/10.1016/j.molcel.2005.12.007
Lalonde M-E, Avvakumov N, Glass KC, Joncas F-H, Saksouk N, Holliday M et al (2013) Exchange of associated factors directs a switch in HBO1 acetyltransferase histone tail specificity. Genes Dev 27:2009–2024. https://doi.org/10.1101/gad.223396.113
Yang X-J (2015) MOZ and MORF acetyltransferases: Molecular interaction, animal development and human disease. Biochim Biophys Acta BBA Mol Cell Res 1853:1818–1826. https://doi.org/10.1016/j.bbamcr.2015.04.014
Su Y, Liu J, Yu B, Ba R, Zhao C (2019) Brpf1 haploinsufficiency impairs dendritic arborization and spine formation, leading to cognitive deficits. Front Cell Neurosci 13:249. https://doi.org/10.3389/fncel.2019.00249
Jd C (2020) The consequences of abnormal gene dosage: lessons from chromosome 18. Trends Genet TIG. https://doi.org/10.1016/j.tig.2020.06.006
Samocha KE, Robinson EB, Sanders SJ, Stevens C, Sabo A, McGrath LM et al (2014) A framework for the interpretation of de novo mutation in human disease. Nat Genet 46:944–950. https://doi.org/10.1038/ng.3050
Savova V, Vinogradova S, Pruss D, Gimelbrant AA, Weiss LA (2017) Risk alleles of genes with monoallelic expression are enriched in gain-of-function variants and depleted in loss-of-function variants for neurodevelopmental disorders. Mol Psychiatry 22:1785–1794. https://doi.org/10.1038/mp.2017.13
Bergendahl LT, Gerasimavicius L, Miles J, Macdonald L, Wells JN, Welburn JPI et al (2019) The role of protein complexes in human genetic disease. Protein Sci 28:1400–1411. https://doi.org/10.1002/pro.3667
You L, Li L, Zou J, Yan K, Belle J, Nijnik A et al (2016) BRPF1 is essential for development of fetal hematopoietic stem cells. J Clin Invest 126:3247–3262. https://doi.org/10.1172/JCI80711
You L, Zou J, Zhao H, Bertos NR, Park M, Wang E et al (2015) Deficiency of the chromatin regulator BRPF1 causes abnormal brain development. J Biol Chem 290:7114–7129. https://doi.org/10.1074/jbc.M114.635250
You L, Chen L, Penney J, Miao D, Yang X-J (2014) Expression atlas of the multivalent epigenetic regulator Brpf1 and its requirement for survival of mouse embryos. Epigenetics 9:860–872. https://doi.org/10.4161/epi.28530
Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D et al (2009) DECIPHER: database of chromosomal imbalance and phenotype in humans using ensembl resources. Am J Hum Genet 84:524–533. https://doi.org/10.1016/j.ajhg.2009.03.010
Rehm HL, Berg JS, Brooks LD, Bustamante CD, Evans JP, Landrum MJ et al (2015) ClinGen–the clinical genome resource. N Engl J Med 372:2235–2242. https://doi.org/10.1056/NEJMsr1406261
Audebert M, Chevillard S, Levalois C, Gyapay G, Vieillefond A, Klijanienko J et al (2000) Alterations of the DNA repair gene OGG1 in human clear cell carcinomas of the kidney. Cancer Res 60:4740–4744
Sampath H, Lloyd RS (2019) Roles of OGG1 in transcriptional regulation and maintenance of metabolic homeostasis. DNA Repair 81:102667. https://doi.org/10.1016/j.dnarep.2019.102667
Reshetnikov VV, Kisaretova PE, Ershov NI, Shulyupova AS, Oshchepkov DY, Klimova NV et al (2020) Genes associated with cognitive performance in the Morris water maze: an RNA-seq study. Sci Rep 10:22078. https://doi.org/10.1038/s41598-020-78997-6
Welch MD, DePace AH, Verma S, Iwamatsu A, Mitchison TJ (1997) The human Arp2/3 complex is composed of evolutionarily conserved subunits and is localized to cellular regions of dynamic actin filament assembly. J Cell Biol 138:375–384. https://doi.org/10.1083/jcb.138.2.375
Xu N, Qu G-Y, Wu Y-P, Lin Y-Z, Chen D-N, Li X-D et al (2020) ARPC4 promotes bladder cancer cell invasion and is associated with lymph node metastasis. J Cell Biochem 121:231–243. https://doi.org/10.1002/jcb.29136
Huang N, Lee I, Marcotte EM, Hurles ME (2010) Characterising and predicting haploinsufficiency in the human genome. PLoS Genet 6:e1001154. https://doi.org/10.1371/journal.pgen.1001154
Fu J, Wang T, Fu Z, Li T, Zhang X, Zhao J et al (2021) Case report: a case report and literature review of 3p deletion syndrome. Front Pediatr. https://doi.org/10.3389/fped.2021.618059
Gunnarsson C, Foyn Bruun C (2010) Molecular characterization and clinical features of a patient with an interstitial deletion of 3p25.3-p26.1. Am J Med Genet A 152A:3110–3114. https://doi.org/10.1002/ajmg.a.33353
Peltekova IT, Macdonald A, Armour CM (2012) Microdeletion on 3p25 in a patient with features of 3p deletion syndrome. Am J Med Genet A 158A:2583–2586. https://doi.org/10.1002/ajmg.a.35559
Acknowledgements
The authors thank Flor Vásquez for her suggestions in our manuscript.
Funding
None.
Author information
Authors and Affiliations
Contributions
HHAB, FCV and RPL interpreted the CMA’s data. HHAB wrote and edited the manuscript. FCV and RPL reviewed the document. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All procedures performed in this study were under the ethical standards of the institutional research committee of Instituto Nacional de Salud del Niño and the 1964 Helsinki Declaration and its later amendments.
Consent for publication
The written informed consent was obtained from the family for this publication.
Competing interests
The authors declare no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Supplemental information about compromised genes in the patient’s CNV and related diseases to date; and frequent clinical manifestations in patients with 3pter-3p25 deletion syndrome.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abarca-Barriga, H.H., Chavesta Velásquez, F. & Punil Luciano, R. Intellectual developmental disorder with dysmorphic facies and ptosis caused by copy number variation including the BRPF1 gene in Peruvian patient. Egypt J Med Hum Genet 23, 141 (2022). https://doi.org/10.1186/s43042-022-00356-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-022-00356-z