
Abstract
Background
It is difficult to classify a small fraction of α- and β-thalassemia (α- and β-thal) carriers based on their Hb A2 levels. Here, we report the results of a molecular investigation in a cohort of thalassemia carriers with borderline Hb A2 levels originated from western Iran.
Results
The documents of 5956 α- or β-thal carriers were reviewed. The frequency of individuals with borderline Hb A2 levels in this cohort was 436 (7.32%). A total of 12 different α-thal and 27 different β-thal variants were identified in this study.
Conclusions
Our data showed that individuals with borderline Hb A2 are not uncommon in our population. Moreover, preselection of α- and β-thal carriers with borderline Hb A2 levels based on Hb A2, mean corpuscular volume (MCV), and mean cell hemoglobin (MCH) is not advisable in our population. Therefore, it is necessary to investigate both α- and β-globin genes in cases with borderline Hb A2 levels, especially if the partner is a carrier of β-thal or α0-thal.
Similar content being viewed by others

Background
Hemoglobin F (Hb F; α2γ2), hemoglobin A2 (Hb A2; α2δ2), and hemoglobin A (Hb A; α2β2) are known as three main types of hemoglobin observed in normal adults [1]. Alpha and beta thalassemia (α- and β-thal) are due to mutations in the alpha (HBA1: hemoglobin subunit alpha1 and HBA2: hemoglobin subunit alpha2) and beta (HBB: hemoglobin subunit beta) genes, respectively [2]. Each normal individual has four α-globin genes [2]. Deletion of one or two α-globin genes leads to α+- or α0-trait in carriers, respectively. Although the carriers of α+-trait are asymptomatic with mildly microcytic and normal levels of Hb A2 (2.4–3.2%), the carriers of α0-trait are characterized by low-to-low normal range of Hb A2 (1.5–2.5%) and slightly reduced in mean corpuscular volume (MCV) and mean cell hemoglobin (MCH) [3]. On the other hand, the Hb A2 level in β-thal carriers is increased to more than 3.5% [4].
Hb A2 quantification is used as a valuable test to differentiate α- and β-thal carriers in clinical laboratories. However, it is difficult to classify a small fraction of thalassemia carriers because of their Hb A2 borderline levels [3]. Regardless of the fact that different studies have considered different ranges of Hb A2 to define the borderline level [3,4,5,6,7,8,9,10,11], subjects with borderline levels of Hb A2 should be carefully screened to avoid missing at-risk couples.
Because thalassemia is a serious issue in the Iranian health system [12,13,14], the National Thalassemia Screening Program has been conducted in Iran for more than 20 years [15]. Implementation of this program in Iran has reduced the birth rate of β-thal by 90.13% [16]. Since 2009, thalassemia carriers from western provinces of Iran including Kermanshah, Kurdistan, Hamadan, Ilam, and Lorestan have been referred to our laboratory (Kermanshah reference Laboratory, Kermanshah, Iran), as one of the designated centers of genetic diagnosis of couples at risk for thalassemia. In this study, we aimed to evaluate the prevalence of subjects with borderline Hb A2 levels among the total population of thalassemia carriers in western Iran and to determine the spectrum and frequency of thalassemia mutations in this group.
Methods
According to the National Thalassemia Screening Program in Iran, marriage registrars refer couples planning to get married to a designated local laboratory for premarital screening. The process of screening has already been discussed elsewhere [15, 17]. In the present study, the results of genetic analysis of 5,956 suspected carriers of thalassemia who were referred to Kermanshah Reference Laboratory, Kermanshah, Iran, between 2009 and 2019 were reviewed. The MCV and/or MCH of these subjects were < 80fL and < 27 pg, respectively. They were originated from five provinces located in the western part of Iran including Kermanshah, Kurdistan, Hamadan, Ilam, and Lorestan (Fig. 1).

Demographic representation of subjects investigated in this study (the map was adapted from https://www.amar.org.ir/)
Using a Capillarys 2 Flex Piercing instrument (Sebia, Lisses, France), the capillary electrophoresis had been performed for all of the subjects, accordingly, they were classified to have borderline (3.1–3.9%) or other Hb A2 levels (< 3.1 or > 3.9) (Fig. 2) [4]. After the collection of peripheral blood samples and genomic DNA extraction using QIAamp DNA Mini kit (Qiagen GmbH, Hilden, Germany), the quality and quantity of extracted DNA were assessed using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). We used three different molecular methods to identify α-thal causing mutations: (1) multiplex gap-polymerase chain reaction (gap-PCR) for detection of—α3.7 (NG_000006.1: g.34164_37967del3804),—α4.2, and—–MED I (NG_000006.1: g.24664_41064del16401) deletions; (2) amplification refractory mutation system (ARMS)-PCR for detection of α–5 nt α (HBA2: c.95+2_95+6delTGAGG), polyadenylation signal A2 or polyA2 (αpolyA2 α, HBA2: c.*92A>G), polyA1 (αpolyA1 α, HBA2:c.*94A>G); ααHb Adana, HBA1: c.179G>A), and αcodon 19 α (HBA2: c.56delG); and (3) direct DNA sequencing of HBA1 and HBA2 genes for the analysis of samples whose mutations were not in the list of mutations examined by the gap-PCR and ARMS-PCR methods. Moreover, ARMS-PCR technique was used for detection of HBB gene mutations. Samples negative on ARMS-PCR were analyzed by direct sequencing of the HBB gene. The protocols, primer sequences, and PCR cycling conditions have been described by us in our previous studies [18,19,20,21,22,23,24,25,26]. Some samples negative on the above-mentioned methods had been sent to Dr. Zeinali’s Medical Genetics Laboratory, Kawsar Human Genetics Research Center, Tehran, Iran, for multiplex ligation-dependent probe amplification (MLPA) analysis of the α- and β-globin gene clusters.

Profile of the study used to evaluate subjects with borderline Hb A2 levels
Statistical data analysis was carried out using the Statistical Package for the Social Sciences software, version 25.0 (https://ibm.com/SPSS-Statistics/). The independent samples t test, the analysis of variance (ANOVA), and Scheffe post hoc analysis were performed to determine the significance of differences between hematological factors in thalassemia genotypes identified in our study. The p values less than 0.05 were considered to be significant for all described tests.
Results
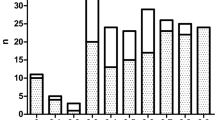
In this study, the documents of 5,956 α- or β-thal carriers were reviewed. The frequency of subjects with borderline Hb A2 levels in this group was 436 (7.32%), in which α- and/or β-variants were identified in 350 subjects (mutation detection rate: 80.28%) (Fig. 2). These subjects classified as carriers with α-thal mutations (n = 134: 38.28%), β-thal mutations (n = 199: 56.86%) and coexistence of α- and β-thal mutations (n = 17: 4.86%), respectively (Fig. 2).
A total of 12 different α-thal and 27 different β-thal mutations were identified in the present study. The most frequent mutations are shown in Table 1 (data not shown in detail). The α-globin mutations including -α3.7 deletion, αPolyA2 α, ααHb Adana, –α4.2 deletion, and α–5 nt α were detected with the frequencies of 62.57, 8.19, 7.02, 5.85 and 5.85%, respectively. On the other hand, Hb D-Punjab (HBB: c.364G >C) was the most frequent β-globin variant (21.92%), followed by IVS-II-1 (G>A) (HBB: c.315+1G>A), IVS-I-6 (T>C) (HBB: c.92+6T>C), codons 8/9 (+G) (HBB: c.27_28insG), IVS-I-110 (G>A) (HBB: c.93-21G>A), –101 (C>T) (HBB: c.-151C>T), Hb Ernz (HBB: c.371C>A), and IVS-I-128 (T>G) (HBB: c.93-3T>G) with the frequencies of 16.89, 13.70, 7.76, 7.31, 4.57, 4.11 and 3.65%, respectively, among our samples.
Out of 59 couples who were at risk for having a child with thalassemia, eight and 51 couples had α-thal and β-thal mutations, respectively. Fifty-two couples were referred for premarital genetic diagnosis, whereas seven couples were referred to us as they already had a child with thalassemia (Table 2). In addition, 37 out of 59 couples were from consanguineous families.
As shown in Table 3, Hb A2, MCV, and MCH were significantly different between α-thal and β-thal carriers (p < 0.0001). Post hoc analysis showed that except for –101 (C>T) and IVS-I-128 (T>G) mutations, the other most prevalent β-thal mutations in this study caused a significant reduction in MCV and MCH levels compared with α-thal mutations (p < 0.05) (Table 4). On the other hand, in contrast to α/- α, αα/αnon-deletionalα (αα/αNDα) or αα/ααND genotypes, there was no statistically significant difference in the mean levels of MCV and MCH factors between β-thal and -α/-α or αα/– genotypes (p > 0.05) (Table 5).
Discussion
Iran is a vast country with high ethnic diversity. These factors have not only led to a high prevalence of a number of genetic diseases, but have also led to a wide range of mutations associated with each of these diseases [27,28,29,30,31,32]. The above issues are more evident in the western region of Iran [19,20,21,22,23,24,25,26, 33,34,35,36,37,38]. In this study, we report the hematological and molecular findings in a group of α- and β-thal carriers with borderline Hb A2 levels (3.1–3.9%). They were originated from western provinces of Iran with Kurd, Lur, and Lak ethnicities.
Various mutations were detected on α- and β-globin genes. As shown in Table 1, the borderline Hb A2 level is not specific to just one or a few α-thal mutations in our population. In addition, all main types of α-thal genotypes including αα/-α, -α/-α, αα/–, αα/αND αα or αα/ααND were associated with borderline Hb A2 levels. This finding is in line with previous studies in which different α-thal mutations were associated with borderline Hb A2 levels [4, 5, 8,9,10, 39, 40]. Previous studies have shown that variations in the erythroid Krüppel-like factor (KLF1) gene lead to a borderline Hb A2 in normal individuals as well as α+- or α0-thal carriers [40, 41]. As shown in Fig. 2, our population has a high frequency of α-thalassemia carriers with borderline Hb A2 levels. Therefore, the KLF1 gene mutations may be a possible cause of increased Hb A2 levels in our population.
Based on the identification of 27 different β-thal mutations associated with borderline Hb A2 in this study (Table 1), it can be concluded that the borderline Hb A2 level is not specific to just one or a few β-thal mutations in our population. Regardless of Hb D-Punjab and Hb Ernz which are known as Hb variants, the IVS-II-1 (G>A), IVS-I-6 (T>C), codons 8/9 (+G), IVS-I-110 (G>A), –101 (C>T) and IVS-I-128 (T>G) were the most frequent HBB gene mutations among our samples. Previous studies have reported that some mild β-thal mutations are recurrently associated with normal/borderline Hb A2 levels. However, their types and frequencies were not similar in different populations. Examples of these mutations are as follows: IVS-I-6 (T>C), –101 (C>T) and IVS-I-110 (G>A) in Italy [8, 10, 42]; cap+1 (A>C) (HBB: c.-50A>C) and IVS-I-5 (G>C) (HBB: c.92+5G>C) in India [6, 43]; IVS-I-6 (T>C), IVS-I-110 (G>A) and IVS-I-1 (G>A) (HBB: c.92+1G>A) in Egypt [44]; and IVS-I-1 (G>A) in Malaysia [7]. All of these mutations were also observed in the present study (Table 2). Therefore, our findings are consistent with the results of the studies mentioned above.
In this study, we found eight couples with α-thal mutations in which one partner had a borderline Hb A2 level (Table 2). According to previous studies [27, 45], there is a probability of 25% of having a child/fetus with Hb H disease or hydrops fetalis in these couples. On the other hand, out of 51 couples with β-thal mutations who were at risk for having a child with thalassemia, we found 40 couples with borderline Hb A2 in one partner and increased Hb A2 level (> 3.9) in the other partner; nine couples with borderline Hb A2 levels in both partners; and two couples with borderline Hb A2 in one partner and normal Hb A2 level in the other partner. In addition, seven couples were referred to us as they already had a child with thalassemia (Table 2).
Our analysis showed that the mean values of Hb A2, MCV, and MCH were significantly different between α-thal and β-thal carriers with borderline Hb A2 levels (p < 0.0001) (Table 3). Two other statistical analyzes were performed. In the first analysis, the mean values of MCV and MCH in β-thal genotypes (as a single group) were compared with those in common β-thal genotypes (Table 4). The results showed that the mean values of these factors were not statistically significant in at least two β-thal genotypes, –101 (C>T)/W and IVS-I-128 (T>G)/W, compared to α-thal genotypes. In the second analysis, the mean values of MCV and MCH were evaluated in β-thal genotypes (as a single group) compared to those in αα/-α, -α/-α, αα/–, αα/αNDα or αα/ααND genotypes (Table 5). According to the results of this analysis, there was no statistically significant difference between the mean values of these factors in -α/-α and αα/– genotypes compared to the β-thal genotypes. In addition, the mean values of MCV and MCH were not significantly different when we compared the combined α-thal and β-thal genotypes with α-thal genotypes (as a single group) or β-thal genotypes (as a single group) (Tables 4, 5, respectively). Therefore, preselection of α- and β-thal carriers based on MCV and MCH factors is not advisable in our population.
Conclusions
In conclusion, our data showed that thalassemia carriers with borderline Hb A2 are not uncommon among all thalassemia carriers in our population. Out of 59 couples who were at risk for having a child with thalassemia, 37 couples were from consanguineous families. In addition, preselection of α- and β-thal carriers with borderline Hb A2 levels based on Hb A2, MCV, or MCH factors, alone or in combination, is not advisable in our population. Therefore, it is necessary to investigate both α- and β-globin genes in cases with borderline Hb A2 levels, especially if couples are from consanguineous families or the partner is a carrier of β-thal or α0-thal. This strategy is cost-effective because it prevents the birth of children with thalassemia and, consequently, the high costs associated with caring of these patients.
Availability of data and materials
Not applicable.
Abbreviations
- Hb A:
-
Hemoglobin A
- Hb A2 :
-
Hemoglobin A2
- Hb F:
-
Hemoglobin F
- α-thal:
-
Alpha thalassemia
- β-thal:
-
Beta thalassemia
- HBA1:
-
Hemoglobin subunit alpha 1
- HBA2:
-
Hemoglobin subunit alpha 2
- HBB:
-
Hemoglobin subunit beta
- MCV:
-
Mean corpuscular volume
- MCH:
-
Mean cell hemoglobin
- ARMS-PCR:
-
Amplification refractory mutation system-polymerase chain reaction
- MLPA:
-
Multiplex ligation-dependent probe amplification
References
Amoyal I, Fibach E (2007) Hemoglobin switch in the newborn: a flow cytometry analysis. Neonatology 91(1):61–68
Angastiniotis M, Lobitz S (2019) Thalassemias: an overview. Int J Neonatal Screen. 5:16
Brancaleoni V, Di Pierro E, Motta I, Cappellini M (2016) Laboratory diagnosis of thalassemia. Int J Lab Hematol 38(Suppl. 1):32–40
Al-Amodi AM, Ghanem NZ, Aldakeel SA, Ibrahim AlAsoom L, Rafique Ahmed N, Almandil NB et al (2018) Hemoglobin A2 (HbA2) has a measure of unreliability in diagnosing β-thalassemia trait (β-TT). Curr Med Res Opin 34(5):945–951
Paglietti ME, Satta S, Sollaino MC, Barella S, Ventrella A, Desogus MF et al (2016) The problem of borderline hemoglobin A2 levels in the screening for β-thalassemia carriers in sardinia. Acta Haematol 135(4):193–199
Gorivale M, Sawant P, Mehta P, Nadkarni A, Ghosh K, Colah R (2015) Challenges in prenatal diagnosis of beta thalassaemia: couples with normal HbA2 in one partner. Prenat Diagn 35(13):1353–1357
Rosnah B, Shahida N, Nazri M, Marini R, Noor HM (2017) The Diagnosis of beta thalassemia with borderline HbA2 level among kelantan population. J Blood Disord Transfus. https://doi.org/10.4172/2155-9864.1000396
Mosca A, Paleari R, Galanello R, Sollaino C, Perseu L, Demartis FR et al (2008) New analytical tools and epidemiological data for the identification of HbA2 borderline subjects in the screening for beta-thalassemia. Bioelectrochemistry 73(2):137–140
Borgio JF, AbdulAzeez S, Al-Muslami AM, Naserullah ZA, Al-Jarrash S, Al-Suliman AM et al (2018) KLF1 gene and borderline hemoglobin A2 in Saudi population. Arch Med Sci 14(1):230–236
Giambona A, Passarello C, Vinciguerra M, Muli RL, Teresi P, Anzà M et al (2008) Significance of borderline hemoglobin A2 values in an Italian population with a high prevalence of β-thalassemia. Haematologica 93(9):1380–1384
Uysal A, Genc A, Taşyürek N, Türkyilmaz B (2013) Prevalence of β-thalassemia trait and abnormal hemoglobin in premarital screening in the Province of Izmir. Turkey Pediatr Hemat Oncol 30(1):46–50
Ehsani MA, Hedayati-Asl AA, Bagheri A, Zeinali S, Rashidi A (2009) Hydroxyurea-induced hematological response in transfusion-independent beta-thalassemia intermedia: case series and review of literature. Pediatr Hemat Oncol 26(8):560–565
Alebouyeh M (2005) Pediatric hematology and oncology in Iran. Pediatr Hemat Oncol 22(1):1–9
Karimi M, Cohan N, De Sanctis V, Mallat NS, Taher A (2014) Guidelines for diagnosis and management of Beta-thalassemia intermedia. Pediatr Hemat Oncol 31(7):583–596
Samavat A, Modell B (2004) Iranian national thalassaemia screening programme. Br Med J 329(7475):1134–1137
Hadipour Dehshal M, Tabrizi Namini M, Hantoushzadeh R, Yousefi DS (2019) β-Thalassemia in Iran: things everyone needs to know about this disease. Hemoglobin 43(3):166–173
Shaianmehr M, Abassi M, Zareipour M, Sadaghianifar A (2016) Premarital screening of beta thalassemia in Urmia county of Northwest Iran during 2014: a short report. J RafsanjanUniv Med Sci 14(10):903–910
Alibakhshi R, Khalegi S, Akramipour R, Bidoki SK (2014) Molecular analysis of alpha globin genes non deletional mutations in alpha thalassemia patients in Kermanshah province. Razi J Med Sci 21(118):13–21
Alibakhshi R, Mehrabi M, Omidniakan L, Shafieenia S (2015) The spectrum of α-thalassemia mutations in Kermanshah Province, West Iran. Hemoglobin 39(6):403–406
Moradi K, Aznab M, Tahmasebi S, Dastafkan Z, Omidniakan L, Ahmadi M et al (2019) The spectrum of α-thalassemia mutations in the Lak population of Iran. Hemoglobin 43(2):107–111
Moradi K, Aznab M, Biglari M, Shafieenia S, Azimi A, Bijari N et al (2020) Molecular genetic analysis of α-thalassemia in Hamadan province. West Iran Hemoglobin 44(5):319–324
Moradi K, Aznab M, Azimi A, Biglari M, Shafieenia S, Alibakhshi R (2020) α-thalassemia mutations in Ilam Province, West Iran. Hemoglobin 19:1–6. https://doi.org/10.1080/03630269.2019.1694033
Alibakhshi R, Moradi K, Aznab M, Dastafkan Z, Tahmasebi S, Ahmadi M et al (2020) The spectrum of α-thalassemia mutations in Kurdistan Province West Iran. Hemoglobin 44(3):156–161
Mehrabi M, Alibakhshi R, Fathollahi S, Farshchi MR (2013) The spectrum of β-thalassemia mutations in Kermanshah Province in West Iran and its association with hematological parameters. Hemoglobin 37(6):544–552
Alibakhshi R, Moradi K, Aznab M, Azimi A, Shafieenia S, Biglari M (2019) The spectrum of β-thalassemia mutations in Hamadan Province, West Iran. Hemoglobin 43(1):18–22
Moradi K, Aznab M, Tahmasebi S, Omidniakan L, Bijari N, Alibakhshi R (2020) Distribution of HBB gene mutations in the Kurdish population of Ilam province, West Iran. Hemoglobin 44(4):244–248
Valaei A, Karimipoor M, Kordafshari A, Zeinali S (2018) Molecular basis of α-thalassemia in Iran. Iran Biomed J 22(1):6–14
Alibakhshi R, Mohammadi A, Khamooshian S, Kazeminia M, Moradi K (2021) CFTR gene mutation spectrum among 735 Iranian patients with cystic fibrosis: a comprehensive systematic review. Pediatr Pulmonol 56(12):3644–3656
Alibakhshi R, Mohammadi A, Salari N, Khamooshian S, Kazeminia M, Moradi K (2021) Spectrum of PAH gene mutations in 1547 phenylketonuria patients from Iran: a comprehensive systematic review. Metab Brain Dis 36:767–780
Bazi A, Miri-Moghaddam E (2016) Spectrum of β-thalassemia mutations in Iran, an Update. Iran J Pediatr Hematol Oncol. 6:3
Mahdieh N, Rabbani B (2016) Beta thalassemia in 31,734 cases with HBB gene mutations: pathogenic and structural analysis of the common mutations; Iran as the crossroads of the Middle East. Blood Rev 30(6):493–508
Alibakhshi R, Mohammadi A, Ghadiri K, Khamooshian S, Kazeminia M, Moradi K (2022) Spectrum of MEFV gene mutations in 4,256 familial Mediterranean fever patients from Iran: a comprehensive systematic review. Egypt J Med Hum Genet 23(1):5
Moradi K, Alibakhshi R, Ghadiri K, Khatami SR, Galehdari H (2012) Molecular analysis of exons 6 and 7 of phenylalanine hydroxylase gene mutations in Phenylketonuria patients in Western Iran. Indian J Hum Genet 18(3):290–293
Moradi K, Alibakhshi R, Khatami S (2013) The proportion of tetrahydrobiopterin deficiency and PAH gene deficiency variants among cases with hyperphenyalaninemia in Western Iran. Indian J Hum Genet 19(4):454–458
Alibakhshi R, Moradi K, Mohebbi Z, Ghadiri K (2014) Mutation analysis of PAH gene in patients with PKU in western Iran and its association with polymorphisms: identification of four novel mutations. Metab Brain Dis 29(1):131–138
Sahami A, Alibakhshi R, Ghadiri K, Sadeghi H (2014) Mutation analysis of exons 10 and 17a of CFTR gene in patients with cystic fibrosis in Kermanshah province, western Iran. J Reprod Infertil 15(1):49–56
Alibakhshi R, Moradi K, Biglari M, Shafieenia S (2018) Spectrum of phenylalanine hydroxylase gene mutations in Hamadan and Lorestan Provinces of Iran and their associations with variable number of tandem repeat alleles. Iran J Med Sci 43(3):318–323
Karimi N, Alibakhshi R, Almasi S (2018) CFTR mutation analysis in Western Iran: identification of two novel mutations. J Reprod Infertil 19(1):3–9
Satta S, Paglietti ME, Sollaino MC, Barella S, Moi P, Desogus MF et al (2017) Changes in HbA2 and HbF in alpha thalassemia carriers with KLF1 mutation. Blood Cell Mol Dis 64:30–32
Yu LH, Liu D, Cai R, Shang X, Zhang XH, Ma XX et al (2015) Changes in hematological parameters in α-thalassemia individuals co-inherited with erythroid Krüppel-like factor mutations. Clin Genet 88(1):56–61
Perseu L, Satta S, Moi P, Demartis FR, Manunza L, Sollaino MC et al (2011) KLF1 gene mutations cause borderline HbA2. Blood J Am Soc Hematol 118(16):4454–4458
Galanello R, Barella S, Ideo A, Gasperini D, Rosatelli C, Paderi L et al (1994) Genotype of subjects with borderline hemoglobin A2 levels: Implication for, β-thalassemia carrier screening. Am J Hematol 46(2):79–81
Rangan A, Sharma P, Dadu T, Saxena R, Verma IC, Bhargava M (2011) β-Thalassemia mutations in subjects with borderline HbA2 values: a pilot study in North India. Clin Chem Lab Med 49(12):2069–2072
Abdel-Messih IY, Youssef SR, Mokhtar GM, Elmogy MI, Mahmoud HM, Ayoub M et al (2015) Clinical to molecular screening paradigm for β-thalassemia carriers. Hemoglobin 39(4):240–246
Hafezi-Nejad N, Khosravi M, Bayat N, Kariminejad A, Hadavi V, Oberkanins C et al (2014) Characterizing a cohort of α-thalassemia couples collected during screening for hemoglobinopathies: 14 years of an iranian experience. Hemoglobin 38(3):153–157
Acknowledgements
We thank all the people at the Medical Genetics Laboratory, Kermanshah University of Medical Sciences, Kermanshah, Iran, for their great kindness and collaboration.
Funding
None.
Author information
Authors and Affiliations
Contributions
RA and KM had the idea of the topic. KM, SS, and AA collected the data in the documents. KM reviewed the data and drafted the manuscript. RA supervised the study process and critically reviewed the manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics Committee of Kermanshah University of Medical Sciences, Kermanshah, Iran (Ethics code: IR.KUMS.REC.1398.731, project number: 980659).
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Moradi, K., Alibakhshi, R., Shafieenia, S. et al. Problem of borderline hemoglobin A2 levels in an Iranian population with a high prevalence of α- and β-thalassemia carriers. Egypt J Med Hum Genet 23, 61 (2022). https://doi.org/10.1186/s43042-022-00279-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-022-00279-9