
Abstract
Background
Chanarin–Dorfman syndrome (CDS; OMIM # 275630) is a rare neutral lipid storage disorder caused by mutation in ABHD5 (a/b hydrolase domain containing 5″) a cofactor for adipose triglyceride lipase (ATGL) resulting in intracellular accumulation of triacylglycerol (TG) in numerous body tissues. It is an autosomal recessive disorder mutation in ABHD5 that causes the partial or total loss of ATGL activation, leading to the accumulation of TG inside lipid droplets. We aim to assess the clinical and biochemical manifestations, diagnosis, follow-up and genotype–phenotype correlations in six Pakistani pediatric patients with CDS.
Results
Six male patients with mean age 15 months (9–24 months) diagnosed as CDS on the basis of non-bullous ichthyosiform erythroderma, hepatomegaly and Jordans bodies in peripheral smear. We identified two novel mutations in ABHD5 gene (c.338G > T and c.730_731insA). These mutations have a pathogenic and damaging influence on the ABHD5 protein structure and function. During the 2 year clinical follow-up one patient died of severe chest infection; he had severe phenotype. There is no genotype–phenotype correlation in CDS. Therapy with low fat diet, MCT oil, Vit E and ursodeoxycholic acid has promising results in CDS.
Conclusion
Non-bullous ichthyosiform erythroderma, steatohepatitis and Jordan’s anomaly are consistent findings in all cases of CDS. It is suggested that an accurate diagnosis of CDS should be based on combination of clinical features and pathognomonic ABHD5 mutations. More studies should be carried out to identify population-specific genetic mutations for the rapid and cost-effective diagnosis of CDS.
Similar content being viewed by others

Background
Chanarin–Dorfman syndrome (CDS; OMIM # 275630) is a rare neutral lipid storage disorder characterized by intracellular accumulation of triacylglycerol (TG) in numerous tissues like skin, liver, central nervous system, ears, eyes and skeletal muscle [1]. It is an autosomal recessive disorder caused by mutation in ABHD5 (α/β hydrolase domain 5), a cofactor for adipose triglyceride lipase (ATGL). Mutations in ABHD5 cause the partial or total loss of ATGL activation, leading to the accumulation of TG inside lipid droplets. These neutral lipid droplets are highly dynamic cellular organelles of eukaryotic cells that provide a rapidly mobilized lipid source for numerous biochemical processes [2].
Clinical symptoms of Chanarin–Dorfman syndrome (CDS) arise from the storage of excess triglycerides including non-bullous ichthyosiform erythroderma, hearing loss, mental retardation, hepatomegaly and myopathy. Non-bullous ichthyosiform erythroderma is the most noticeable finding of the CDS, is thought to be due to the impairment of ABDH5 function to activate PNPLA1, which catalyzes the final step of \({\acute{\omega}}\)–O–acylceramide production, an essential lipid for the permeability of skin barrier [3]. Diagnosis of CDS is based on clinical findings and the presence of lipid vacuoles in granulocytes in Giemsa staining of peripheral blood smear known as Jordan’s anomaly [4]. Out of one hundred and twenty-eight CDS reported patients only 85 of them have been confirmed by ABHD5 genetic analysis [5].
We present the clinical presentation, diagnosis, treatment and follow-up of six Pakistani CDS patients along their molecular analysis. We observed two novel homozygous pathogenic mutations in our cohort.
Methods
This study was performed in accordance with institutional review board standards. All patients were included in the study, with non-bullous congenital ichthyosiform erythroderma syndrome and diagnosed on the basis of clinical symptoms and biochemical parameters of CDS. Their data are shown in Table 1. The written informed consent was taken from patient’s parents.
Molecular analysis
Blood samples of affected children were drawn in EDTA vials and stored at − 4 °C until used for further process. For genomic analysis DNA extraction was carried out by using modified phenol chloroform method [6]. Double stranded DNA was used to enrich target regions from fragmented genomic DNA with the Twist Human Core Exome Plus kit. The generated library is sequenced on an Illumina platform to obtain at least 20× coverage depth for > 98% of the targeted bases. The ABHD5 sequence was taken from ENSEMBL Database, and primers were designed using Primer 3 v. 0.4.0. An inhouse bioinformatics pipeline, including read alignment to GRCh37/hg19 genome assembly, variant calling (single nucleotide and small deletion/insertion variants), annotation and comprehensive variant filtering were applied. The investigations for relevant variants were focused on coding exons and flanking ± 20 intronic nucleotides of genes.
In silico analysis
To ascertain the potential biological impact of the observed missense variant on Abhd5 function, four widely used online predictors were employed: SIFT (http://sift.jcvi.org/), Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/), Mutation taster (http://www.mutationtaster.org/), PROVEAN (http://provean.jcvi.org/) and Mutation Assessor Release 2 (www.mutationassessor.org/). Multiple sequence alignment (MSA) was performed using theMEGA7 software [7]. The protein model was made using the iTaser server for protein prediction structure and function prediction to estimate the crystal structure of human ABHD5 protein bound with ligands.
Results
Demographic and clinical data
Case 1
A 9-month-old boy presented with progressive abdominal distension for 3 months. He was born at term through uneventful vaginal delivery. He achieved gross motor and fine motor milestones in the form of eye to eye contact, social smile, neck holding, sitting and standing with support appropriately. He was also having dry scaly skin and was on dermatologist follow-up. His clinical examination at the time of diagnosis is shown in Table 1. Now he is 2.5 years of age doing well on low fat diet, Medium chain triglyceride (MCT) oil, ursodeoxycholic acid, and Vitamin E and skin emollients (Fig. 2 for pedigree).
Case 2
He was born preterm (34 weeks) via vaginal delivery with low birth weight at private clinic of lady health worker (no record available); at birth he had ichthyotic skin over hands and dorsum of feet. He neither achieved his fine motor skills in the form of eye to eye contact, social smile nor gross motor skills like neck holding and sitting. At 5 months of age he developed generalized ichthyosis over face, limbs, abdomen and back. At 11 months of age he presented to pediatric gastrointestinal clinic for his chronic diarrhea and failure to thrive. He had global developmental delay, bilateral convergent squint, hypotonia and microcephaly. His liver histopathology and peripheral smear is shown in Fig. 1a–c. He expired at 18 months of age due to severe lower respiratory tract infection (Fig. 2 for pedigree).

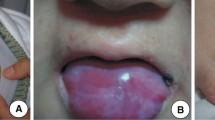
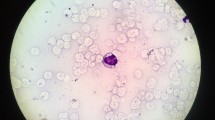
Liver histology and Jordan’s anomaly in a CDS patient (case 2). (a) Liver parenchyma (H&E) shows distorted lobular architecture and expanded portal areas. Trichrome stain reveals portal and periportal fibrosis along with many thin fibrous septa suggestive of steatohepatitis with fibrosis. (b) Higher magnification shows hepatocytes are enlarged with compressed sinusoids and severe pan lobular steatosis. (c) Microphotographs of May–Grünwald–Giemsa buffy coats showing Jordan’s anomaly

Pedigrees
Case 3
A 13-month-old male child presented with Ichthyosis since 7th day of life. Progressive abdominal distension noticed by parents since 9 months of life. He achieved all his fine motor and gross motor milestones appropriately like neck holding at 4 months of age, sitting with support at 8 months of age, standing at 13 months. He can walk independently, has achieved one sentence speech and is partial toilet trained. His skin manifestations are shown in Fig. 3a and b He is now 2 years and 7 months of age and spending healthy life. He is taking low fat diet, Medium chain triglyceride (MCT oil, ursodeoxycholic acid, Vitamin E and skin emollients (Fig. 2 for pedigree).

Dermatological characterization of CDS patients. Lamellar ichthyosis affecting facial region, trunk and extremities of Case 3 (a, b), Case 5 (c, d)
Case 4
A 16-month-old boy born by consanguineous parents (consanguinity for all patients is drawn in Fig. 2) was referred to pediatric liver clinic for his hepatomegaly and deranged liver functions tests. He was found to have generalized ichthyosis. He was clinically diagnosed as Chanarin–Dorfman syndrome by the presence of Jordan anomaly. His liver biopsy revealed steatohepatitis. Currently he is 3 years of age, and his liver functions are improved by using low fat diet, Medium chain triglyceride (MCT) oil, ursodeoxycholic acid and Vitamin E.
Case 5
A one-and-half-year-old male child presented with hepatomegaly at liver clinic at children hospital and the institute of child health Lahore. He was first born child of consanguineous (Fig. 2 for pedigree) parents and was found to have ichthyosis since birth (Fig. 3c, d). For which he was on follow-up of a dermatologist. He was developmentally normal vaccinated child.
On examination he was found to have generalized dry scaly skin (Fig. 3). Abdomen was soft, distended with normal umbilicus. Liver was palpable 4 cm below right subcostal margin, total span 10 cm firm in consistency and rounded margins. Spleen was not palpable. No evidence of free fluid in peritoneum was observed. All other systemic examination including cardiovascular, respiratory and central nervous system was normal. His laboratory data are shown in Table 1. His buffy coat smear confirms Jordan anomaly (Fig. 1c). Currently he is three and half years old, doing well on low fat diet, Medium chain triglyceride (MCT) oil, ursodeoxycholic acid, and Vitamin E and skin emollients.
Case 6
A 2-year-old boy was brought by his mother for his dry scaly skin involving lower limbs only. She was concerned for his nutritional status. He was found to have hepatomegaly and deranged lipid profile. He was clinically diagnosed as CDS by presence of Jordan anomaly on peripheral blood smear (Fig. 2 for pedigree).
Sequencing of ABHD5 gene
Sequencing of ABHD5 gene led to the identification of two homozygous novel mutations. Genetic analysis of patient 6 revealed a novel substitution of G into T at c.338 nucleotide position in exon 3. Both parents were heterozygous (carrier) for this variation. The homozygous substitution replaced a codon GGA for Glycine (G) with a valine (V) GTA at 113 codon. The novel missense variant (c.338G > T or p.113G > V) was found to be probably damaging, as predicted by PolyPhen-2 and other tools (Tables 2 and 3). Different online in silico tools were used to determine the evolutionary conservation of the wild amino acid (glycine), three dimensional structures and to predict the effect that mutation has on the protein confirmation.
Sequence analysis of patients 1,2,3,4 and 5 revealed an insertion of A at position 730 in exon 5. c.730_731 Ins A (p.T244Nfs*10) resulted in replacement of a codon ACT for threonine to AAC for asparagine amino acid at codon 244. As a consequence of frameshift, a premature stop codon appears in the translated region and causes shortening of the protein from the normal 349–253 amino acids. The resulting truncated protein is susceptible to non-sense-mediated decay (Table 2). Both parents of all patients (case1-5) were found to be heterozygous (carrier) for this variant.
The c.338G > T missense mutation and c.730_731 Ins A insertion variation was identified as a novel mutation upon confirmation in the general population. For this purpose, 100 healthy controls were taken from the general population and were sequenced for both of these variants. Sequence analysis showed that no such variations were observed in a single control.
Discussion
All of our patients had major clinical symptoms of Chanarin–Dorfman syndrome (CDS) [8] including non-bullous congenital ichthyosiform erythroderma, hepatomegaly, liver steatosis and Jordan’s bodies.
Serum lipid profile was deranged in all of our patients but the level of serum triglycerides (TGs) had been inconsistent with clinical manifestation. Case 2 had mildly elevated TGs but severe disease with markedly elevated liver transaminases, severe steatohepatitis, CNS involvement and myopathy. Other patients had markedly elevated triglyceride levels and very low-density lipoproteins with mild clinical manifestation. Sano et al. demonstrated that the severity of ichthyosis positively correlates with TG level in the scales from patients [9].
Hepatic involvement is variable in CDS, ranging from hepatomegaly and liver steatosis to cirrhosis [10]. Although two of our patients (1 and 3) had liver function tests within normal range, liver biopsy showed steatohepatitis in all patients, and case 2 had bridging fibrosis too. This is consistent with the literature reporting that liver of 64% cases is clinically affected and histological steatohepatitis is found in the 100% CDS patients [11]. Myopathy was present in one of our patients (Case 2), electromyographic examination (EMG) revealed a myopathic pattern and elevation in CPK levels even at 11 month of age contradicting reports that reveal it usually appears late [12]. Same patient has global developmental delay, bilateral convergent squint and pale optic disks but his hearing was normal.
To date more than 42 mutations have been reported in HGMD Professional 2019.4 (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=ABHD5). We found two Novel c.338G > T (p.G113V) and c.730_731insA (p.T244Nfs*10) mutations in the ABHD5 gene. Both of these mutations were detected in the heterozygous state in both parents of all patients.
To confirm the missense mutation and insertion in ABHD5, reported in this study, different in-silico tools were used to evaluate the effects of these variants on the ABHD5 protein structure and function. Pathogenicity predictions by SIFT, PolyPhen-2, Mutation Taster, Provean and Mutation Assessor programs unanimously indicated that these variants are deleterious. It has been established that highly conserved amino acid sequences have functional value and are important for the protein structure, which suggests that they play a key role in determining the conformation of different domains of a protein. Multiple sequence alignment of ABHD5 orthologues and phylogenetic tree analysis showed that the amino acid glycine at position 113 and threonine at 244 in the ABHD5 protein are highly conserved across species, from human to Zebra fish. The evolutionary conservation of this amino acid shows its importance in the structure of the ABHD5 protein (Fig. 4a, b), indicating that amino acid change at this position will significantly affect the ABHD5 protein structure and function. The predicted effects of these variants agree well with the clinical condition/outcome observed in these patients.

a Phylogenetic tree shows the conservation of Abhd5 protein in other orthologs (across different species).The evolutionary history was inferred using the Neighbor–Joining method [1]. The optimal tree with the sum of branch length = 1.01547038 is shown. The evolutionary distances were computed using the Poisson correction method. b Multiple sequence alignment of Abhd5 protein showing high conservation of amino acids across the mutated region of c.730–731insA (p.Thr244Asnfs*10) variation among different species. Mutation position marked in black box
The c.338G > T (p.G113V) mutation is located within a domain, annotated in UniProt as AB hydrolase-1. The mutation introduces an amino acid with different properties, which can disturb this domain and abolish its function. The wild-type residue is a glycine, the most flexible of all residues. This flexibility might be necessary for the protein's function. Mutation of glycine at this position can abolish ABHD5 protein function. The glycine at 113th position is located on the surface of the protein; mutation of this residue can disturb interactions with other molecules or other parts of the protein. The torsion angles for this residue are unusual. Only glycine is flexible enough to make these torsion angles; mutation into another residue will force the local backbone into an incorrect conformation and will disturb the local structure (Fig. 5a, b).

Structure of protein Comparison of the structure of Abhd5 normal protein (a) with mutated protein (b)
The c.730_731InsA (p.Thr244Asnfs*10) insertion resulted in replacement of threonine to asparagine amino acid at codon 244. As a consequence, a premature stop codon appears in the translated region and causes shortening of the protein from the normal 349–253 amino acids. The resulting truncated protein is susceptible to non-sense-mediated decay (Fig. 5c).
No genotype phenotype correlation has been documented in CDS in literature [13]. This is confirmed in our study; five of our patients (1, 2, 3, 4 and 5) had same novel homozygous mutation (c.730_731insA (p.T244Nfs*10)) in the ABHD5. The patient (case2) had a severe phenotype with markedly elevated transaminases and liver fibrosis, myopathy and Central nervous system involvement.
We followed up our patients for 2 years on fat restricted diet, MCT oil, vitamin E and ursodeoxycholic acid. Five of our patients (1, 3, 4, 5, and 6) showed improvement in serum TGs, liver transaminases, liver span and growth centiles. They did not develop any new symptoms like muscle or central nervous system involvement. The skin manifestation required skin emollients and the scaly ichthyotic changes still persisting. We hypothesize that skin manifestation may take longer to revert back and need long-term follow-up.
Conclusion
In conclusion, we describe two novel mutations in ABHD5 gene (c.338G > T and c.730-731insA) in six Pakistani families along with 2 year follow-up. These mutations have a pathogenic and damaging influence on the ABHD5 protein structure and function. Non-bullous ichthyosiform erythroderma, steatohepatitis and Jordan’s anomaly are consistent findings in all cases of CDS.
There is no genotype–phenotype correlation in CDS. Therapy with low fat diet, MCT oil, Vit E and ursodeoxycholic acid has promising results in CDS. It is suggested that an accurate diagnosis of CDS should be based on combination of clinical features and pathognomonic ABHD5 mutations. More studies should be carried out to identify population-specific genetic mutations for the rapid and cost-effective diagnosis of CDS.
Availability of data and materials
Yes Data will be made available on demand.
Abbreviations
- ABHD5:
-
α/β Hydrolase domain
- ALT:
-
Alanine aminotransferase
- ALP:
-
Alkaline phosphatase
- AST:
-
Aspartate aminotransferase
- ATGL:
-
Adipose triglyceride lipase
- CDS:
-
Chanarin–Dorfman syndrome
- CPK:
-
Creatine phosphor kinase
- CNS:
-
Central nervous system
- DNA:
-
Deoxyribonucleic acid
- ENSEMBL:
-
Ensembl genome database project
- EDTA:
-
Ethylene diamine tetra acetic acid
- GGT:
-
Gamma glutamyl-transferase
- HCT:
-
Hematocrit
- HDL:
-
High-density lipoprotein
- HB:
-
Hemoglobin
- INR:
-
International normalization ratio
- LDL:
-
Low-density lipoprotein
- L:
-
Lymphocytes
- OFC:
-
Occipito-frontal circumference
- OMIM:
-
Online Mendelian inheritance in man
- MCT:
-
Medium chain triacylglycerol
- MCV:
-
Mean corpuscular volume
- MCH:
-
Mean corpuscular hemoglobin
- N:
-
Neutrophils
- STB:
-
Serum total bilirubin
- TLC:
-
Total leukocyte count
- TG:
-
Triacylglycerol
- VLDL:
-
Very low-density lipoprotein
- Vit E:
-
Vitamin E
References
Schweiger M, Lass A, Zimmermann R, Eichmann TO, Zechner R (2009) Neutral lipid storage disease: genetic disorders caused by mutations in adipose triglyceride lipase/PNPLA2 or CGI-58/ABHD5. Am J Physiol Endocrinol Metab 297:289–296
Missaglia S, Coleman RA, Mordente A, Tavian D (2019) Neutral lipid storage diseases as cellular model to study lipid droplet function. Cells 8(2):187. https://doi.org/10.3390/cells8020187
Eskiocak AH, Missaglia S, Moro L et al (2019) A novel mutation of ABHD5 gene in a Chanarin Dorfman patient with unusual dermatological findings. Lipids Health Dis 18:232. https://doi.org/10.1186/s12944-019-1181
Tavian D, Colombo R (2007) Improved cytochemical method for detecting Jordans’ bodies in neutral-lipid storage diseases. J Clin Pathol 60:956–958
Durdu M, Missaglia S, Moro L, Tavian D (2018) Clinical and genetic characterization of a Chanarin Dorfman syndrome patient born to diseased parents. BMC Med Genet 19:1–5
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, New York
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Ujihara M, Nakajima K, Yamamoto M, Teraishi M, Uchida Y, Akiyama M et al (2010) Epidermal triglyceride levels are correlated with severity of ichthyosis in Dorfman–Chanarin syndrome. J Dermatol Sci 57:102–107
Redaelli C, Coleman RA, Moro L, Dacou-Voutetakis C, Elsayed SM, Prati D, Colli A, Mela D, Colombo R, Tavian D (2010) Clinical and genetic characterization of Chanarin–Dorfman syndrome patients: first report of large deletions in the ABHD5 gene. Orphanet J Rare Dis 5:33
Takeichi T, Sugiura K, Tso S, Simpson MA, McGrath JA, Akiyama M (2016) Bi-allelic nonsense mutations in ABHD5 underlie a mild phenotype of Dorfman–Chanarin syndrome. J Dermatol Sci 81:134–136
Bruno C, Bertini E, Di Rocco M, Cassandrini D, Ruffa G, De Toni T et al (2008) Clinical and genetic characterization of Chanarin–Dorfman syndrome. Biochem Biophys Res Commun 369:1125–1128
Pennisi EM, Arca M, Bertini E, Bruno C, Cassandrini D, Damico A et al (2017) clinical/genetic features and natural history in a large cohort of Italian patients. Orphanet J Rare Dis 12:90
Nur BG, Gencpinar P, Yuzbasioglu A, Emre SD, Mihci E (2015) Chanarin Dorfman syndrome: genotype-phenotype correlation. Eur J Med Genet 58:238–242
Acknowledgements
The authors are grateful to the patients for their kind cooperation and Prof. Roberto Colombo, head of center for the study of rare inherited disease (CeSMER) Catholic University of the Sacred Heart, Milan, Italy and Centogene the rare disease company Germany for Genome Analysis for exome sequencing.
Funding
None.
Author information
Authors and Affiliations
Contributions
NW conceived, designed and did statistical analysis, writing and editing of manuscript and was the main contributor. SI analyzed and interpreted molecular data of novel and known variant. ZF performed histological examination of liver biopsy and hematological analysis of peripheral blood smear. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical review committee of Pakistan institute of medical sciences, Pakistan, has consented for this study Ref # No.F1-1/2015/ERB/SZABMU/646 dated 14-09-2020 and written informed consent has been taken from parents and Guardians of children included in the study for sharing of data and pictures.
Consent for publication
The parents of patients have consented to publish data including pictures of their children.
Competing interests
None.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Waheed, N., Ijaz, S. & Fayyaz, Z. Chanarin–Dorfman syndrome: clinical/genetic features and natural history in six Pakistani patients. Egypt J Med Hum Genet 22, 69 (2021). https://doi.org/10.1186/s43042-021-00189-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-021-00189-2