
Abstract
Background
Ankylosing spondylitis (AS) is a rare inflammatory disorder affecting the spinal joints. Although we know some of the genetic factors that are associated with the disease, the molecular basis of this illness has not yet been fully elucidated, and the genes involved in AS pathogenesis have not been entirely identified. The current study aimed at constructing a gene network that may serve as an AS gene signature and biomarker, both of which will help in disease diagnosis and the identification of therapeutic targets. Previously published gene expression profiles of 16 AS patients and 16 gender- and age-matched controls that were profiled on the Illumina HumanHT-12 V3.0 Expression BeadChip platform were mined. Patients were Portuguese, 21 to 64 years old, were diagnosed based on the modified New York criteria, and had Bath Ankylosing Spondylitis Disease Activity Index scores > 4 and Bath Ankylosing Spondylitis Functional Index scores > 4. All patients were receiving only NSAIDs and/or sulphasalazine. Functional enrichment and pathway analysis were performed to create an interaction network of differentially expressed genes.
Results
ITM2A, ICOS, VSIG10L, CD59, TRAC, and CTLA-4 were among the significantly differentially expressed genes in AS, but the most significantly downregulated genes were the HLA-DRB6, HLA-DRB5, HLA-DRB4, HLA-DRB3, HLA-DRB1, HLA-DQB1, ITM2A, and CTLA-4 genes. The genes in this study were mostly associated with the regulation of the immune system processes, parts of cell membrane, and signaling related to T cell receptor and antigen receptor, in addition to some overlaps related to the IL2 STAT signaling, as well as the androgen response. The most significantly over-represented pathways in the data set were associated with the “RUNX1 and FOXP3 which control the development of regulatory T lymphocytes (Tregs)” and the “GABA receptor activation” pathways.
Conclusions
Comprehensive gene analysis of differentially expressed genes in AS reveals a significant gene network that is involved in a multitude of important immune and inflammatory pathways. These pathways and networks might serve as biomarkers for AS and can potentially help in diagnosing the disease and identifying future targets for treatment.
Similar content being viewed by others


Background
Ankylosing spondylitis (AS) is a rheumatic disorder of the axial skeleton that is characterized by inflammation and osteoproliferation, leading to a gradual loss of spinal mobility [1]. First identified in the late 1600s, AS is a type of spondyloarthropathy that predominantly affects young men, with a higher prevalence among white Europeans and a lower prevalence among sub-Saharan Africans [2,3,4].
AS is a highly heritable condition, and familial history has been strongly correlated with its pathogenesis in a number of twin studies [5,6,7]. Since 1973, the HLA-B27 gene, a major histocompatibility complex (MHC) class I allele, has been recognized as a major genetic risk factor for AS [8]. In fact, HLA-B27 frequency in a certain population is correlated with the prevalence of spondyloarthropathies, including AS, among its individuals [9, 10]. However, the disease is far from being monogenic in nature. In fact, the more than 100 genetic variants associated with AS account for no more than 30% of its heritability, indicating that much research needs to be carried out in this regard [11].
Despite being associated with several environmental and genetic factors, AS genetics and pathogenesis are poorly understood. The main aim of the current study was to improve the understanding of AS on the genetic level. Thus, in the present study, publicly available gene expression profiles of AS patients were analyzed using several bioinformatics techniques. A gene network was constructed that may serve as an AS gene signature for disease diagnosis and therapeutic target identification.
Methods
Data acquisition
The dataset used in the current study was obtained from The National Center for Biotechnology Information’s (NCBI) Gene Expression Omnibus (GEO) repository (accession number GSE25101) [12].
It is worthwhile to note here that this was the only dataset available for ankylosing spondylitis at the time of writing this paper. The data contained peripheral blood samples from active AS patients (n = 16) and gender- and age-matched controls (n = 16) that were profiled on the Illumina HumanHT-12 V3.0 Expression BeadChip platform. As per the original study by Pimentel-Santos et al., total RNA was extracted from whole blood samples according to standard PAXgene protocol, PAXgene Blood RNA System® tubes (Qiagen, Doncaster, VIC, Australia), was quantified and the integrity assessed by Agilent 2100 BioAnalyser (Agilent, Santa Clara, CA, USA). Only samples with RNA integrity number above 7.5 were used [12].
Patients were Portuguese, aged 21 to 64, and had Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) scores of > 4 and Bath Ankylosing Spondylitis Functional Index (BASFI) scores of > 4. They were diagnosed using the updated New York criteria. Patients were receiving NSAIDs and/or sulphasalazine only. No patients treated with TNF, corticosteroids, or methotrexate were included [12]. For further details of the study subjects and complete microarray processing, please refer to the study by Pimentel-Santos et al. [12].
Data analysis and pre-processing
Over 10,000 genes were identified as differentially expressed (DE) genes after using the GEO2R software that is available on NCBI’s website (https://www.ncbi.nlm.nih.gov/geo/geo2r/). GEO2R facilitates the R-based analysis of GEO data and presents it as a list of genes that is sorted by significance [13]. Further case-control comparison analysis of this gene list was carried out using Microsoft Excel in order to identify the most DE genes in AS, yielding a total of 114 genes. After applying a strict p-value cut-off (p < 0.05), the final gene list consisted of 97 genes, 5 genes were removed for lack of identifier, and hence the final cured list of genes was 92. One of the genes was the HLA gene locus that contained 6 genes, and these 6 genes were each entered separately instead of a one locus. The final gene list contained 97 genes which was used for the final analysis and network construction.
Functional enrichment analysis
Following that, the 97 genes were entered into three databases for gene-gene network and pathway analysis: the Molecular Signatures Database (MSigDB), the geneMANIA database, and the Reactome Pathway database.
The MSigDB is a collection of annotated gene sets where a predefined gene set can be examined for overlap with the previously annotated genes in this database https://www.gsea-msigdb.org/gsea/msigdb/index.jsp. It is used with the Gene set enrichment analysis software (GSEA). GSEA is an analytical software that extracts insights from RNA expression analysis using gene sets, which are groups of genes with common function, location, or regulation (https://www.gsea-msigdb.org/gsea/index.jsp). For this first tool, the 97 genes were compared for overlap with three main gene set categories in MSigDB, the Gene Ontology (GO), Hallmark collection, and the canonical pathways (CP).
Similarly, GeneMANIA is an online tool that is used to predict the function of poorly understood genes as well as the genes that are most likely to be functionally similar (http://genemania.org/). For this second tool, the top 20 DE genes were input, in addition to the genes that were most downregulated (fold change < 0.6 and p-value < 0.05) and the genes that were most upregulated (fold change > 1.24 and p-value < 0.05).
For the next tool, Reactome which is an open-source and peer-reviewed database that facilitates stable over-representation analysis of genome-wide datasets (https://reactome.org/) [14,15,16,17,18], the full list of 97 genes were input for analysis.
Lastly, the Search Tool for the Retrieval of Interacting Genes (STRING) database, v. 11.0, was used to analyze high-confidence (> 0.7) protein-protein network interactions among the 97 DE genes [19]. The STRING database scores protein-protein interaction information from public sources and complements this data with computational predictions (https://string-db.org/).
Results
Identification of differentially expressed (DE) genes
Scrutinization of the 10,000 DE genes yielded 97 genes with a significant level of DE. All genes were deemed significant based on the parametric p-value only, as none were significant in terms of the adjusted p-value. Genes with no identifier were not included in the analysis. The most significantly DE genes were the ITM2A, ICOS, VSIG10L, CD59, TRAC, and CTLA-4 genes, among others (Supplementary file 1). However, the most significantly downregulated genes (fold change [FC] < 0.5 and p-value < 0.05) were the HLA histocompatibility gene locus: HLA-DRB6, HLA-DRB5, HLA-DRB4, HLA-DRB3, HLA-DRB1, HLA-DQB1, the integral membrane protein 2A gene (ITM2A), and cytotoxic T-lymphocyte associated protein 4 gene (CTLA-4).
Pathway analysis (MSigDB overlaps)
Figure 1 illustrates the number of genes in each of the results of the overlap with the MSigDB genesets. Tables 1, 2, and 3 specify the genes in the overlaps. Namely, overlaps were most common for the CD3E, IL2RA, CD27, CTLA-4, and ICOS genes. It is interesting to note here that the top overlapping genes were associated with regulation of the immune system processes, parts of cell membrane, and signaling related to T cell receptor and antigen receptor, in addition to some overlaps related to the IL2 STAT signaling, as well as the androgen response.

Analysis of the overlap of the cured genes with the three annotated sets in MsigDB Gene ontology BP and CC (GO), b Hallmark, and c canonical pathways (CP). The y-axis depicts the number of overlapping genes and the x-axis lists the pathway terms. Blue indicates cellular component (CC); red, biological process (BP); orange, Hallmark; and green, canonical pathways (CP)
Gene network analysis (GeneMania)
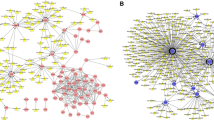
Three gene networks were constructed: one for the top 20 DE genes (Fig. 2), one for the most significantly downregulated genes (fold change < 0.6 and p-value < 0.05) (Fig. 3), and one for the genes that were most upregulated (fold change > 1.24 and p-value < 0.05) (Fig. 4). For the top 20 DE genes, the genes had 57.5% co-expression, 29.91% physical interactions, 7.58% co-localization. 2.51% of the gens had predicted interactions, 2.01% had shared protein domains, and only 0.49% were part of a pathway. For the downregulated genes, they had 78.04% physical interactions, 7.3% co-localization. 7.27% of the gens had predicted interactions, 4.95% were part of a pathway, 1.68% had genetic interactions, and 0.67% shared protein domains. Finally, for the upregulated genes, they had 67.64% physical interactions, 13.5% co-expression, 6.35% were predicted, 6.17% had co-localization, 4.35% were part of a pathway, 1.4% had genetic interactions, and 0.59% had shared protein domains. Functions of the genes are presented in each figure.

Construction of a gene-gene network using the top 20 genes with the most significant differential expression. Black circles with white stripes indicate genes that were entered as query terms, while black circles indicate the associated genes. The size of the circle corresponds with the number of correlations with other genes in the network

Construction of a gene-gene network using the most significantly downregulated genes (fold change < 0.6 and p-value < 0.05). Black circles with white stripes (inside circle) indicate genes that were entered as query terms, while black circles indicate the associated genes. The size of the circle corresponds with the number of correlations with other genes in the network

Construction of a gene-gene network using the most significantly upregulated genes (fold change > 1.24 and p-value < 0.05). Black circles with white stripes (inside circle) indicate genes that were entered as query terms, while black circles indicate the associated genes. The size of the circle corresponds with the number of correlations with other genes in the network
REACTOME pathway investigation
The most significantly over-represented pathways in the AS samples were the “RUNX1 and FOXP3 which control the development of regulatory T lymphocytes (Tregs)” and the “GABA receptor activation” pathways (Fig. 5).

Results of query of Reactome. Top 25 most over-represented pathways in terms of the false discovery rate (FDR)
Protein-protein interaction (PPI) network
The STRING database, v. 11.0, was utilized to identify high-confidence protein-protein interactions (PPI) of the most DE genes. With a significantly low PPI enrichment value (4.83e−6), it can be ascertained that the submitted query items are at least partially biologically connected as a group (Fig. 6).

High-confidence protein-protein interactions of the significantly differentially expressed genes. Disconnected network nodes are not shown
Discussion
HLA-B*27 is a major susceptibility allele for ankylosing spondylitis (AS), with over 95% of AS patients expressing this genotype. However, AS heritability is not fully understood, as the vast majority of individuals with the HLA-B*27 genotype do not develop AS [20]. An interplay between genetics and the environment is believed to be involved in AS, and its environmental trigger is thought to be ubiquitous [5]. In addition, the HLA-B*27 accounts for only around 20% of AS heritability, indicating the need for further scientific elucidation [20, 21]. In this study, we identified several DE genes, upregulated genes, and downregulated genes in AS. The significantly downregulated genes (p < 0.05; FC < 0.6) in AS were namely several HLA class II genes, the ITM2A gene, and the CTLA-4 gene.
The most downregulated genes in AS in the present study were HLA-DQ B1 and HLA-DR (B1, B3, B4, B5) genes as well as the HLA-DRB6 pseudogene. These genes were previously related to the inheritance in multiple sclerosis. For instance, epistasis between the HLA-DRB1 and HLA-DQB1 alleles was found to determine the susceptibility to multiple sclerosis [22]. The HLA-DQB1 gene is responsible for exogenous peptide presentation and encodes one of the two components of the HLA-DQ molecule, which regulates T cell response to islet cell autoantigens [23, 24]. In fact, HLA-DQB1 alleles have been identified as susceptibility loci for type I diabetes, celiac disease, multiple sclerosis, and narcolepsy [22, 25,26,27,28]. With regard to AS, HLA-DQB1 was previously associated with the age of onset and severity of AS in a Han Chinese population [29]. However, in Tunisian AS patients, HLA-DRB1 and HLA-DQB1 were not directly linked to the disease [30]. Among HLA-B27-negative AS patients, negative associations were observed for the HLA-DRB1*15:01, HLA-DQB1*02:01, and HLA-DQB1*06:02 alleles, but a positive association was seen for the HLA-DRB1*11 allele [31]. In one large previous study, there was an independent association with variants in the HLA-A, HLA-DPB1, and HLA-DRB1 loci with AS [32]. It is likely that the role of these genes in AS is more complex and population related, and we must keep in mind that our dataset is derived from a Portuguese population.
The second most downregulated gene was the integral membrane protein 2A (ITM2A) gene. ITM2A is primarily involved in the cellular differentiation of chondrocytes, myocytes, and osteoblasts as well as cellular autophagy [33,34,35]. In fact, enhanced ITM2A expression inhibits the chondrogenic differentiation of mesenchymal stem cells [36]. Also, ITM2A downregulation has been associated with decreased autophagy of breast and ovarian cancer cells [37, 38]. ITM2A was reported to be consistently downregulated in a meta-analysis of four AS case-control datasets [39]. Our results confirm this.
The cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) gene, also known as CD152, was the third most significantly downregulated gene in AS patients. This gene encodes the CTLA-4 surface glycoprotein, an inhibitory molecule that is constitutively expressed by T cells and which maintains tolerance to self-antigens [40]. Although the precise mechanisms are not fully understood, interactions of ligands with CTLA-4 serve to inhibit T cell responses [41]. Klocke et al. created a murine model for CTLA-4 deficiency by breeding mice in which CTLA-4 can be conditionally deleted in adulthood using tamoxifen treatment. The mice developed abnormal immune activation, multiorgan lymphocyte infiltration, and autoantibodies in a short period of time [42]. CTLA-4 polymorphisms have been significantly associated with autoimmune disorders, namely AS, Graves’ disease, and rheumatoid arthritis, in British Caucasian, Irish Caucasian, Iranian, West Algerian, and West Mexican populations but not in a Turkish population [43,44,45,46,47]. It is interesting that the HLA genes and the CTLA-4 proteins were connected through the CD3E protein (Fig. 6).
In the present study, the genes with the most overlaps in AS patients were the CD3E, IL2RA, CD27, CTLA-4, ICOS, ITGA6, SERPINE2, and CD2 genes. The CD3E gene encodes a part of the T cell receptor-CD3 (TCR/CD3) complex, which is essential in linking antigen recognition to signal transduction. CD3E has been previously associated with type I diabetes in women, but it did not show a significant association with susceptibility to coeliac disease [48, 49]. Moreover, a reduced number of immunoreceptor tyrosine-based activation motifs (ITAMs) in CD3E, among the other TCR/CD3 complex subunits, was associated with a breakdown in central tolerance in a murine model [50]. To our knowledge, CD3E was not previously related to AS.
Similarly, the interleukin 2 receptor subunit alpha (IL2RA) gene, also known as CD25, encodes a part of the IL-2 receptor, a cytokine signaling molecule that is important for microbial defense and self-recognition. In one study, IL2RA polymorphisms did not confer the risk of acute anterior uveitis with AS, while another study found that the IL2RA rs2104286 polymorphism was associated with intermediate uveitis but not HLA-B27-associated acute anterior uveitis [51, 52]. Transcriptome analysis identified IL2RA as a potential candidate gene for AS, but genotyping assays found no association between IL2RA polymorphisms and non-anterior uveitis susceptibility [53].
The CD27 gene encodes a tumor necrosis factor (TNF) receptor that is thought to act as a co-stimulatory immune checkpoint for T cell generation and maintenance [54]. The CD70 molecule belongs to the tumor necrosis factor ligand superfamily, and its transient availability on lymphocytes and dendritic cells activates B or T cells that express CD27 [55, 56]. As part of a surface ligand-receptor pair with CD70, CD27 dysregulation has been linked to inflammatory and autoimmune disease [57]. Clonally expanded T cells belonged to the CD27−/CD28− populations in 2 AS patients, while 18 newly diagnosed AS patients were found to exhibit a decreased frequency of CD27+ B cells compared to controls [58, 59].
The inducible T cell co-stimulator (ICOS) gene, also known as CD278, is part of the CD28 superfamily receptors, along with CD28 and CTLA-4. ICOS is essential for T cell function and activation, and it is expressed on activated CD4 and CD8 T cells [60, 61]. It was discovered to have important roles in T cell proliferation and cytokine secretion after it was first identified as a marker of T cell activation [62]. ICOS superinduces IL10 and co-induces a number of other cytokines [63,64,65]. ICOS dysregulation has been previously implicated in a murine model of spondyloarthritis, where it was found to be essential to maintaining the balance between IL10 and IL17 expression [66]. B cells express the ICOS ligand (ICOSL) constitutively, which is inducible in monocytes, dendritic cells, fibroblasts, and endothelial cells. Blocking of the ICOS-ICOSL interactions inhibits both T-helper (Th) type 2 and Th1-dependent responses [60]. In an AS context, conflicting findings were reported with regard to CD4+/CXCR5+/ICOS+ T cell percentages in the peripheral blood of AS patients [67,68,69,70].
The integrin alpha-6 (ITGA6) gene, which encodes a transmembrane receptor, mediates adhesion on the cell-to-cell and cell-to-matrix levels, and it is commonly upregulated in a wide range of tumors [71]. In fact, inhibition of the ITGA6 gene led to the suppression of tumor angiogenesis and gallbladder cancer growth [72]. ITGA6 has been previously identified as part of the differential expression profile of rheumatoid arthritis, and it was among the most differentially expressed genes in a murine model of AS [73, 74]. Similarly, the CD2 gene encodes a cell adhesion molecule that is expressed on the surfaces of natural killer cells and T cells as well as some sub-populations of normal B cells [75]. In rheumatoid arthritis, high levels of CD2 activation epitope T11-3 were reported on blood and synovial fluid T cells [76].
The serpin family E member 2 (SERPINE2) gene encodes a glycoprotein that is expressed on a diverse range of cell types, but it has a key role in the pathophysiology of cardiac fibrosis [77]. Little information is available on the role of the SERPINE2 gene in AS, but, in osteoarthritis, this gene has been reported to prevent cartilage catabolism via the inhibition of MMP-13 expression [78].
There are some limitations to the present study. Genes were only found to be significantly expressed when considering the parametric p-value and not the adjusted p-value. Moreover, the analyzed dataset was obtained from a Portuguese population, which might limit the extrapolation of these findings to other populations. We could assume that these patients are ethnically homogenous, as the study by Pimentel-Santos did, but we also do not have information about other diseases that these patients may have had that may have interfered with the results. Thus, the results of our analysis need to be further validated in a different set of patients and their matched controls. Since the research was retrospective in nature, we were unable to collect any additional information about the patients or the microarray characteristics. This research discovered many new genes and networks. However, further studies may be needed to validate the effect of these genes/networks in a clinical setting.
Conclusion
We have described the differential expression profile of AS patients from a previously published dataset using recent analysis techniques. Our data pointed to the involvement of immune system processes and inflammatory processes, as well as other processes and pathways that were not described in the original article. These pathways and networks once validated might serve as biomarkers for AS and can potentially help in diagnosing the disease and identifying future targets for treatment.
Availability of data and materials
This study used a published dataset for its analysis. The dataset used in the current study was obtained from The National Center for Biotechnology Information’s (NCBI) Gene Expression Omnibus (GEO) repository (accession number GSE25101) [12].
Abbreviations
- AS:
-
Ankylosing spondylitis
- BASDAI:
-
Bath Ankylosing Spondylitis Disease Activity Index
- BASFI:
-
Bath Ankylosing Spondylitis Functional Index
- CP:
-
Canonical pathways
- DE:
-
Differentially expressed
- GEO:
-
Gene Expression Omnibus
- GO:
-
Gene Ontology
- GSEA:
-
Gene Set Enrichment Analysis software
- MSigDB:
-
Molecular Signatures Database
- NSAIDs:
-
Nonsteroidal anti-inflammatory drugs
- NCBI:
-
National Center for Biotechnology Information
- STRING:
-
Search Tool for the Retrieval of Interacting Genes
References
Braun J, Sieper J (2007) Ankylosing spondylitis. Lancet. 369(9570):1379–1390. https://doi.org/10.1016/S0140-6736(07)60635-7
Dean LE, Jones GT, MacDonald AG, Downham C, Sturrock RD, Macfarlane GJ (2014) Global prevalence of ankylosing spondylitis. Rheumatology. 53(4):650–657. https://doi.org/10.1093/rheumatology/ket387
Braun J, Bollow M, Remlinger G, Eggens U, Rudwaleit M, Distler A et al (1998) Prevalence of spondylarthropathies in HLA-B27 positive and negative blood donors. Arthritis Rheum 41(1):58–67. https://doi.org/10.1002/1529-0131(199801)41:1<58::AID-ART8>3.0.CO;2-G
Lee W, Reveille JD, Davis JC, Learch TJ, Ward MM, Weisman MH (2007) Are there gender differences in severity of ankylosing spondylitis? Results from the PSOAS cohort. Ann Rheum Dis 66(5):633–638. https://doi.org/10.1136/ard.2006.060293
Brown MA, Kennedy LG, MacGregor AJ, Darke C, Duncan E, Shatford JL et al (1997) Susceptibility to ankylosing spondylitis in twins: the role of genes, HLA, and the environment. Arthritis Rheum 40(10):1823–1828. https://doi.org/10.1002/art.1780401015
JÄRvinen P. (1995) Occurrence of ankylosing spondylitis in a nationwide series of twins. Arthritis Rheum 38(3):381–383. https://doi.org/10.1002/art.1780380313
Morin M, Hellgren K, Frisell T (2020) Familial aggregation and heritability of ankylosing spondylitis - a Swedish nested case-control study. Rheumatol (United Kingdom) 59(7):1695–1702
Brewerton DA, Hart FD, Nicholls A, Caffrey M, James DCO, Sturrock RD (1973 Apr) Ankylosing spondylitis and HL-A 27. Lancet. 301(7809):904–907. https://doi.org/10.1016/S0140-6736(73)91360-3
Sheehan NJ. The ramifications of HLA-B27. Vol. 97, J R Soc Med Royal Society of Medicine Press; 2004. p. 10–14.
Stolwijk C, Boonen A, van Tubergen A, Reveille JD (2012) Epidemiology of spondyloarthritis. Rheum Dis Clin North Am NIH Public Access 38:441–476
Ranganathan V, Gracey E, Brown MA, Inman RD, Haroon N (2017) Pathogenesis of ankylosing spondylitis-recent advances and future directions. Nat Rev Rheumatol 13(6):359–367. https://doi.org/10.1038/nrrheum.2017.56
Pimentel-Santos FM, Ligeiro D, Matos M, Mourão AF, Costa J, Santos H et al (2011) Whole blood transcriptional profiling in ankylosing spondylitis identifies novel candidate genes that might contribute to the inflammatory and tissue-destructive disease aspects. Arthritis Res Ther 13(2)
Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M et al (2013) NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res 41(D1):D991–D995
Jassal B, Matthews L, Viteri G, Gong C, Lorente P, Fabregat A et al (2020) The reactome pathway knowledgebase. Nucleic Acids Res 48(D1):D498
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102(43):15545–15550. https://doi.org/10.1073/pnas.0506580102
Zuberi K, Franz M, Rodriguez H, Montojo J, Lopes CT, Bader GD, Morris Q (2013) GeneMANIA Prediction Server 2013 update. Nucleic Acids Res 41(W1):W115–W122. https://doi.org/10.1093/nar/gkt533
Fabregat A, Sidiropoulos K, Viteri G, Forner O, Marin-Garcia P, Arnau V, D’Eustachio P, Stein L, Hermjakob H (2017) Reactome pathway analysis: a high-performance in-memory approach. BMC Bioinformatics 18(1):142. https://doi.org/10.1186/s12859-017-1559-2
Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP (2011) Molecular signatures database (MSigDB) 3.0. Bioinformatics. 27(12):1739–1740. https://doi.org/10.1093/bioinformatics/btr260
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, Jensen LJ, Mering C (2019) STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 47(D1):D607–D613. https://doi.org/10.1093/nar/gky1131
Zhu W, He X, Cheng K, Zhang L, Chen D, Wang X et al (2019) Ankylosing spondylitis: etiology, pathogenesis, and treatments. Bone Res Sichuan University 7
Breban M, Said-Nahal R, Hugot JP, Miceli-Richard C (2003) Familial and genetic aspects of spondyloarthropathy. Rheum Dis Clin North Am WB Saunders 29:575–594
Lincoln MR, Ramagopalan SV, Chao MJ, Herrera BM, Deluca GC, Orton SM et al (2009) Epistasis among HLA-DRB1, HLA-DQA1, and HLA-DQB1 loci determines multiple sclerosis susceptibility. Proc Natl Acad Sci U S A 106(18):7542–7547. https://doi.org/10.1073/pnas.0812664106
Tree TIM, Duinkerken G, Willemen S, De Vries RRP, Roep BO (2004) HLA-DQ-regulated T-cell responses to islet cell autoantigens insulin and GAD65. Diabetes. 53(7):1692–1699. https://doi.org/10.2337/diabetes.53.7.1692
Whyte JM, Ellis JJ, Brown MA, Kenna TJ (2019) Best practices in DNA methylation: lessons from inflammatory bowel disease, psoriasis and ankylosing spondylitis. Vol. 21, Arthritis Research and Therapy. BioMed Central Ltd:1–14
Horn GT, Bugawan TL, Long CM, Erlich HA (1988) Allelic sequence variation of the HLA-DQ loci: relationship to serology and to insulin-dependent diabetes susceptibility. Proc Natl Acad Sci U S A 85(16):6012–6016. https://doi.org/10.1073/pnas.85.16.6012
Todd JA, Bell JI, McDevitt HO (1987) HLA-DQβ gene contributes to susceptibility and resistance to insulin-dependent diabetes mellitus. Nature. 329(6140):599–604. https://doi.org/10.1038/329599a0
Miyagawa T, Toyoda H, Kanbayashi T, Imanishi A, Sagawa Y, Kotorii N, Kotorii T, Hashizume Y, Ogi K, Hiejima H, Kamei Y, Hida A, Miyamoto M, Ikegami A, Wada Y, Takami M, Fujimura Y, Tamura Y, Omata N, Masuya Y, Kondo H, Moriya S, Furuya H, Kato M, Kojima H, Kashiwase K, Saji H, Khor SS, Yamasaki M, Ishigooka J, Wada Y, Chiba S, Yamada N, Okawa M, Kuroda K, Kume K, Hirata K, Uchimura N, Shimizu T, Inoue Y, Honda Y, Mishima K, Honda M, Tokunaga K (2015) An association analysis of HLA-DQB1 with narcolepsy without cataplexy and idiopathic hypersomnia with/without long sleep time in a Japanese population. Hum Genome Var 2(1):15031. https://doi.org/10.1038/hgv.2015.31
Murray JA, Moore SB, van Dyke CT, Lahr BD, Dierkhising RA, Zinsmeister AR, Melton LJ III, Kroning CM, el–Yousseff M, Czaja AJ (2007) HLA DQ gene dosage and risk and severity of celiac disease. Clin Gastroenterol Hepatol 5(12):1406–1412. https://doi.org/10.1016/j.cgh.2007.08.013
Hao J, Liu Y, Xu J, Wang W, Wen Y, He A, Fan Q, Guo X, Zhang F (2017) Genome-wide DNA methylation profile analysis identifies differentially methylated loci associated with ankylosis spondylitis. Arthritis Res Ther 19(1):177. https://doi.org/10.1186/s13075-017-1382-1
Kchir MM, Hamdi W, Laadhar L, Kochbati S, Kaffel D, Saadellaoui K, Lahmar H, Ghannouchi MM, Azzouz D, Daoud L, Ben Hamida A, Zouari B, Zitouni M, Makni S (2010) HLA-B, DR and DQ antigens polymorphism in Tunisian patients with ankylosing spondylitis (a case-control study). Rheumatol Int 30(7):933–939. https://doi.org/10.1007/s00296-009-1079-0
Reveille JD, Zhou X, Lee MJ, Weisman MH, Yi L, Gensler LS, Zou H, Ward MM, Ishimori ML, Learch TJ, He D, Rahbar MH, Wang J, Brown MA (2019) HLA class I and II alleles in susceptibility to ankylosing spondylitis. Ann Rheum Dis 78(1):66–73. https://doi.org/10.1136/annrheumdis-2018-213779
Cortes A, Pulit SL, Leo PJ, Pointon JJ, Robinson PC, Weisman MH, Ward M, Gensler LS, Zhou X, Garchon HJ, Chiocchia G, Nossent J, Lie BA, Førre Ø, Tuomilehto J, Laiho K, Bradbury LA, Elewaut D, Burgos-Vargas R, Stebbings S, Appleton L, Farrah C, Lau J, Haroon N, Mulero J, Blanco FJ, Gonzalez-Gay MA, Lopez-Larrea C, Bowness P, Gaffney K, Gaston H, Gladman DD, Rahman P, Maksymowych WP, Crusius JBA, van der Horst-Bruinsma IE, Valle-Oñate R, Romero-Sánchez C, Hansen IM, Pimentel-Santos FM, Inman RD, Martin J, Breban M, Wordsworth BP, Reveille JD, Evans DM, de Bakker PIW, Brown MA (2015) Major histocompatibility complex associations of ankylosing spondylitis are complex and involve further epistasis with ERAP1. Nat Commun 6(1):7146. https://doi.org/10.1038/ncomms8146
Namkoong S, Lee KI, Lee JI, Park R, Lee EJ, Jang IS, Park J (2015) The integral membrane protein ITM2A, a transcriptional target of PKA-CREB, regulates autophagic flux via interaction with the vacuolar ATPase. Autophagy [Internet] 11(5):756–768 Available from: http://www.tandfonline.com/doi/full/10.1080/15548627.2015.1034412
Deleersnijder W, Hong G, Cortvrindt R, Poirier C, Tylzanowski P, Pittois K et al (1996) Isolation of markers for chondro-osteogenic differentiation using cDNA library subtraction. Molecular cloning and characterization of a gene belonging to a novel multigene family of integral membrane proteins. J Biol Chem [Internet] 271(32):19475–19482 Available from: http://www.jbc.org/
Plas D, Merregaert J (2004) In vitro studies on Itm2a reveal its involvement in early stages of the chondrogenic differentiation pathway. Biol Cell 96(6):463–470. https://doi.org/10.1016/j.biolcel.2004.04.007
Boeuf S, Börger M, Hennig T, Winter A, Kasten P, Richter W (2009) Enhanced ITM2A expression inhibits chondrogenic differentiation of mesenchymal stem cells. Differentiation. 78(2–3):108–115. https://doi.org/10.1016/j.diff.2009.05.007
Zhou C, Wang M, Yang J, Xiong H, Wang Y, Tang J (2019) Integral membrane protein 2A inhibits cell growth in human breast cancer via enhancing autophagy induction. Cell Commun Signal [Internet] 17(1):105 Available from: https://biosignaling.biomedcentral.com/articles/10.1186/s12964-019-0422-7
Nguyen TMH, Shin IW, Lee TJ, Park J, Kim JH, Park MS, Lee EJ (2016) Loss of ITM2A, a novel tumor suppressor of ovarian cancer through G2/M cell cycle arrest, is a poor prognostic factor of epithelial ovarian cancer. Gynecol Oncol 140(3):545–553. https://doi.org/10.1016/j.ygyno.2015.12.006
Lee YH, Choi SJ, Ji JD, Song GG (2014) THU0494 meta-analysis of differentially expressed genes in ankylosing spondylitis. Ann Rheum Dis 73(Suppl 2):354.2-354. https://doi.org/10.1136/annrheumdis-2014-eular.1187
Romo-Tena J, Gómez-Martín D, Alcocer-Varela J (2013) CTLA-4 and autoimmunity: new insights into the dual regulator of tolerance. Autoimmun Rev Elsevier 12:1171–1176
Rowshanravan B, Halliday N, Sansom DM (2018) Europe PMC Funders Group CTLA-4 : a moving target in immunotherapy. Blood. 131(1):58–67, CTLA-4: a moving target in immunotherapy. https://doi.org/10.1182/blood-2017-06-741033
Klocke K, Sakaguchi S, Holmdahl R, Wing K (2016) Induction of autoimmune disease by deletion of CTLA-4 in mice in adulthood. Proc Natl Acad Sci U S A 113(17):E2383–E2392. https://doi.org/10.1073/pnas.1603892113
Dahmani CA, Benzaoui A, Amroun H, Mecabih F, Sediki FZ, Zemani-Fodil F, Fodil M, Boughrara W, Mecheti B, Attal N, Mehtar N, Petit-Teixeira E, Boudjema A (2018) Association of the HLA-B27 antigen and the CTLA4 gene CT60/rs3087243 polymorphism with ankylosing spondylitis in Algerian population: a case–control study. Int J Immunogenet 45(3):109–117. https://doi.org/10.1111/iji.12369
Karakose Okyaltırık F, Rezvani A, Turan S, Uysal Ö, Yakar F, Sozgen H, Ozkan NE, Akkoyunlu ME, Bayram M, Kutbay Ozcelik H, Sezer M, Guler M, Yaylım İ, Kart L (2017) Relationship of CTLA4 and CD28 polymorphisms with lung involvement, HRCT findings and pulmonary function tests in Turkish patients with ankylosing spondylitis. Clin Respir J 11(5):593–601. https://doi.org/10.1111/crj.12388
Azizi E, Massoud A, Amirzargar AA, Mahmoudi M, Soleimanifar N, Rezaei N, Jamshidi AR, Nikbin B, Nicknam MH (2010) Association of CTLA4 gene polymorphism in iranian patients with ankylosing spondylitis. J Clin Immunol 30(2):268–271. https://doi.org/10.1007/s10875-009-9356-y
Vaidya B, Imrie H, Perros P, Young ET, Kelly WF, Carr D, Large DM, Toft AD, McCarthy M, Kendall-Taylor P, Pearce SH (1999) The cytotoxic T lymphocyte antigen-4 is a major Graves’ disease locus. Hum Mol Genet 8(7):1195–1199. https://doi.org/10.1093/hmg/8.7.1195
García-Chagollán M, Ledezma-Lozano IY, Hernández-Bello J, Sánchez-Hernández PE, Gutiérrez-Ureña SR, Muñoz-Valle JF (2020) Expression patterns of CD28 and CTLA-4 in early, chronic, and untreated rheumatoid arthritis. J Clin Lab Anal 34(5)
Wong S, Moore S, Orisio S, Millward A, Demaine AG (2008) Susceptibility to type I diabetes in women is associated with the CD3 epsilon locus on chromosome 11. Clin Exp Immunol 83(1):69–73. https://doi.org/10.1111/j.1365-2249.1991.tb05590.x
Brophy K, Ryan AW, Turner G, Trimble V, Patel KD, O’Morain C, et al. Evaluation of 6 candidate genes on chromosome 11q23 for coeliac disease susceptibility: a case control study. BMC Med Genet 2010;11(1):1–8, DOI: https://doi.org/10.1186/1471-2350-11-76.
Holst J, Wang H, Eder KD, Workman CJ, Boyd KL, Baquet Z, Singh H, Forbes K, Chruscinski A, Smeyne R, van Oers NSC, Utz PJ, Vignali DAA (2008) Scalable signaling mediated by T cell antigen receptor-CD3 ITAMs ensures effective negative selection and prevents autoimmunity. Nat Immunol [Internet] 9(6):658–666. Available from: https://www.nature.com/articles/ni.1611. https://doi.org/10.1038/ni.1611
Yu H, Liu Y, Zhang L, Wu L, Zheng M, Cheng L, Luo L, Kijlstra A, Yang P (2014) FoxO1 gene confers genetic predisposition to acute anterior uveitis with ankylosing spondylitis. Investig Ophthalmol Vis Sci 55(12):7970–7974. https://doi.org/10.1167/iovs.14-15460
Lindner E, Weger M, Steinwender G, Griesbacher A, Posch U, Ulrich S, Wegscheider B, Ardjomand N, el-Shabrawi Y (2011) IL2RA gene polymorphism rs2104286 A>G seen in multiple sclerosis is associated with intermediate uveitis: Possible parallel pathways? Investig Ophthalmol Vis Sci 52(11):8295–8299. https://doi.org/10.1167/iovs.11-8163
Zhu Z-Q, Tang J-S, Cao X-J (2013) Transcriptome network analysis reveals potential candidate genes for ankylosing spondylitis. Eur Rev Med Pharmacol Sci 17(23):3178–3185
Hendriks J, Gravestein LA, Tesselaar K, Van Lier RAW, Schumacher TNM, Borst J (2000) CD27 is required for generation and long-term maintenance of T cell immunity. Nat Immunol 1(5):433–440. https://doi.org/10.1038/80877
Wajant H (2016) Therapeutic targeting of CD70 and CD27 [Internet]. Expert Opin Ther Targets Taylor and Francis Ltd 20:959–973 [cited 2021 Mar 12], Available from: https://www.tandfonline.com/doi/abs/10.1517/14728222.2016.1158812
Borst J, Hendriks J, Xiao Y (2005) CD27 and CD70 in T cell and B cell activation. Curr Opin Immunol 17(3):275–281. https://doi.org/10.1016/j.coi.2005.04.004
Han BK, Olsen NJ, Bottaro A (2016) The CD27-CD70 pathway and pathogenesis of autoimmune disease. Semin Arthritis Rheum WB Saunders 45:496–501
Mamedov IZ, Britanova OV, Chkalina AV, Staroverov DB, Amosova AL, Mishin AS, Kurnikova MA, Zvyagin IV, Mutovina ZY, Gordeev AV, Khaidukov SV, Sharonov GV, Shagin DA, Chudakov DM, Lebedev YB (2009) Individual characterization of stably expanded T cell clones in ankylosing spondylitis patients. Autoimmunity. 42(6):525–536. https://doi.org/10.1080/08916930902960362
Niu XY, Zhang HY, Liu YJ, Zhao D, Shan YX, Jiang YF (2013 Nov) Peripheral B-cell activation and exhaustion markers in patients with ankylosing spondylitis. Life Sci 93(18–19):687–692. https://doi.org/10.1016/j.lfs.2013.09.003
Guillonneau C, Aubry V, Renaudin K, Séveno C, Usal C, Tezuka K, Anegon I (2005) Inhibition of chronic rejection and development of tolerogenic T cells after ICOS-ICOSL and CD40-CD40L co-stimulation blockade. Transplantation. 80(2):255–263. https://doi.org/10.1097/01.TP.0000165429.57421.D6
Wikenheiser DJ, Stumhofer JS (2016) ICOS co-stimulation: friend or foe? Front Immunol Frontiers Media SA 7:1
Metzger TC, Long H, Potluri S, Pertel T, Bailey-Bucktrout SL, Lin JC, Fu T, Sharma P, Allison JP, Feldman RMR (2016) ICOS promotes the function of CD4+ effector T cells during anti-OX40-mediated tumor rejection. Cancer Res 76(13):3684–3689. https://doi.org/10.1158/0008-5472.CAN-15-3412
Hutloff A, Dittrich AM, Beier KC, Eljaschewitsch B, Kraft R, Anagnostopoulos I, Kroczek RA (1999) ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature. 397(6716):263–266. https://doi.org/10.1038/16717
Dong C, Juedes AE, Temann UA, Shresta S, Allison JP, Ruddle NH, Flavell RA (2001) ICOS co-stimulatory receptor is essential for T-cell activation and function. Nature. 409(6816):97–101. https://doi.org/10.1038/35051100
Beier KC, Hutloff A, Dittrich AM, Heuck C, Rauch A, Bchner K et al (2000) Induction, binding specificity and function of human ICOS. Eur J Immunol 30(12):3707–3717. https://doi.org/10.1002/1521-4141(200012)30:12<3707::AID-IMMU3707>3.0.CO;2-Q
Araujo LM, Fert I, Jouhault Q, Labroquère K, Andrieu M, Chiocchia G, Breban M (2014) Increased production of interleukin-17 over interleukin-10 by Treg cells implicates inducible costimulator molecule in experimental spondyloarthritis. Arthritis Rheum 66(9):2412–2422. https://doi.org/10.1002/art.38737
Wu S, Yang T, Pan F, Xia G, Hu Y, Liu L, Fan D, Duan Z, Ding N, Xu S, Cai G, Wang L (2014) Increased frequency of circulating follicular helper T cells in patients with ankylosing spondylitis. Mod Rheumatol 25(1):110–115. https://doi.org/10.3109/14397595.2014.902149
Xiao F, Zhang HY, Liu YJ, Zhao D, Shan YX, Jiang YF (2013) Higher frequency of peripheral blood interleukin 21 positive follicular helper T cells in patients with ankylosing spondylitis. J Rheumatol 40(12):2029–2037. https://doi.org/10.3899/jrheum.130125
Long S, Ma L, Wang D, Shang X (2018) High frequency of circulating follicular helper T cells is correlated with B cell subtypes in patients with ankylosing spondylitis. Exp Ther Med 15(5):4578–4586. https://doi.org/10.3892/etm.2018.5991
Bautista-Caro M-B, Arroyo-Villa I, Castillo-Gallego C, de Miguel E, Peiteado D, Plasencia-Rodríguez C, et al. Decreased frequencies of circulating dollicular helper T cell counterparts and plasmablasts in ankylosing spondylitis patients naïve for TNF blockers. Szodoray P, editor. PLoS One. 2014;9(9):e107086.
Brooks DLP, Schwab LP, Krutilina R, Parke DN, Sethuraman A, Hoogewijs D, Schörg A, Gotwald L, Fan M, Wenger RH, Seagroves TN (2016) ITGA6 is directly regulated by hypoxia-inducible factors and enriches for cancer stem cell activity and invasion in metastatic breast cancer models. Mol Cancer 15(1):26. https://doi.org/10.1186/s12943-016-0510-x
Jin YP, Hu YP, Wu XS, Wu YS, Ye YY, Li HF et al (2018) MiR-143-3p targeting of ITGA6 suppresses tumour growth and angiogenesis by downregulating PLGF expression via the PI3K/AKT pathway in gallbladder carcinoma. Cell Death Dis 9(2):1–15
Ma H, Xu D, Fu Q (2012) Identification of ankylosing spondylitis-associated genes by expression profiling. Int J Mol Med 30(3):693–696. https://doi.org/10.3892/ijmm.2012.1047
Chen YJ, Chang WA, Wu LY, Hsu YL, Chen CH, Kuo PL (2018) Systematic analysis of differential expression profile in rheumatoid arthritis chondrocytes using next-generation sequencing and bioinformatics approaches. Int J Med Sci 15(11):1129–1142. https://doi.org/10.7150/ijms.27056
Kingma DW, Imus P, Xie XY, Jasper G, Sorbara L, Stewart C, Stetler-Stevenson M (2002) CD2 is expressed by a subpopulation of normal B cells and is frequently present in mature B-cell neoplasms. Cytometry. 50(5):243–248. https://doi.org/10.1002/cyto.10131
Potocnik AJ, Menninger H, Yang SY, Pirner K, Krause A, Burmester GR et al (1991) Expression of the CD2 activation epitope T11-3 (CD2R) on T cells in rheumatoid arthritis, juvenile rheumatoid arthritis, systemic lupus urythematosus, ankylosing spondylitis, and lyme disease: phenotypic and functional analysis. Scand J Immunol 34(3):351–358. https://doi.org/10.1111/j.1365-3083.1991.tb01556.x
Li X, Zhao D, Guo Z, Li T, Qili M, Xu B et al (2016) Overexpression of SerpinE2/protease nexin-1 contribute to pathological cardiac fibrosis via increasing collagen deposition. Sci Rep 6(1):1–12
Santoro A, Conde J, Scotece M, Abella V, Lois A, Lopez V, et al. SERPINE2 inhibits IL-1α-induced MMP-13 expression in human chondrocytes: involvement of ERK/NF-κB/AP-1 pathways. d’Acquisto F, editor. PLoS One. 2015;10(8):e0135979.
Acknowledgements
The authors would like to acknowledge the efforts of the University of Petra’s Faculty of Pharmacy and Medical Sciences, its Dean, and its Department Head of Pharmacology and Biomedical Sciences.
Funding
This study was not funded.
Author information
Authors and Affiliations
Contributions
RA: Conceptualization, review and editing of manuscript, and analysis validation and visualization. AK: Formal analysis, methodology, and writing original draft. AT: visualization and final manuscript review and editing. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The full list of gene names and abbreviations is provided in the supplementary data.
Supplementary Information
Additional file 1: Table 1.
List of submitted genes converted into NCBI (Entrez) genes through the MSigDB.
Additional file 2.
List of DE genes downloaded from GEO2R.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
AlEjielat, R., Khaleel, A. & Tarkhan, A.H. Differential gene expression analysis of ankylosing spondylitis shows deregulation of the HLA-DRB, HLA-DQB, ITM2A, and CTLA4 genes. Egypt J Med Hum Genet 22, 42 (2021). https://doi.org/10.1186/s43042-021-00161-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-021-00161-0