
Abstract
Background
The X-linked myotubular myopathy (XLMTM) is a rare congenital disease. Its main symptoms are hypotonia, dysmorphic facial features, respiratory failure, and feeding disorder.
Case presentation
This study reports on a male patient from Neonatal Intensive Care Unit, who presented symptoms of congenital myopathy. After eliminating many other possible causes, he was eventually proven to bear a c.197C>G, p.(Thr66Arg) MTM1 mutation, a variant of uncertain significance, never described in the literature before. Family of the patient underwent the same genetic tests that proved the mother to be the carrier of mutation.
Conclusion
The article is a first report on abovementioned, newly discovered mutation in MTM1 gene, with high probability leading to the centronuclear myopathy phenotype. It also summarizes the diagnostic process and current state of knowledge about the therapy and prognosis for children with XLMTM. The authors hope that the findings will contribute to the diagnostic process of subsequent patients.
Similar content being viewed by others

Background
Centronuclear myopathy is a group of congenital diseases characterized by muscle weakness and centrally located nuclei in the muscle fibers [1]. Its type is dependent on genomic mutation present in the patient. One of the genes responsible for the development of the disease is MTM1. It is located in the chromosomal region Xq28 and encodes a protein named myotubularin [2]. Deleterious changes in MTM1 gene trigger the absence or loss of the function of myotubularin, a lipid phosphatase involved in electrical stimuli translation in the neuromuscular junction [3, 4]. These variants are inherited in a X-linked manner, resulting in a subtype of centronuclear myopathy named X-linked myotubular myopathy (XLMTM). Its incidence is estimated for approximately 1 in 50,000 newborn males [5], whereas the penetrance reaches 100% [6]. The phenotype varies from mild, that does not reduce the survivability, to the most often, classic severe form with few patients surviving into adulthood [6, 7]. The typical phenotype consists of neonatal hypotonia, respiratory distress, high-arched palate, elongated face, premature adrenarche, cryptorchidism, ophthalmoplegia, pyloric stenosis, and hepatic peliosis [8]. To date, more than 250 pathological variants of MTM1 have been published in the medical literature [9,10,11,12]. To our knowledge, none of them was described as c.197C>G, p.(Thr66Arg). This mutation also has not been mentioned in the disease-related variation databases as Clin-Var or HGMD (Human Gene Mutation Database). We report on a male patient from Neonatal Intensive Care Unit that presented with typical symptoms of a congenital myopathy and after a thorough diagnostic process was proven to bear a c.197C>G, p.(Thr66Arg) MTM1 mutation, described as a variant of uncertain significance (VUS).
Case presentation
A male neonate in his first day of life was admitted to the Neonatal Intensive Care Unit (NICU) because of the respiratory failure, significant diffuse muscle weakness, and hypotonia (Fig. 1). He was born in the 39th week of pregnancy by caesarean section. The neonate presented with apnea and bradycardia and was intubated in the first minute of life. He scored 2/4/4/4 Apgar points after 1/3/5/10 min of life, respectively. A few dysmorphic features were clearly visible: dolichocephaly, prominent forehead, bitemporal narrowing, hypertelorism, up-turned nose, tent-shaped upper lip, high arched palate, micrognatia, low-set ears, and pectus excavatum were present (Figs. 2, 3, and 4). Moreover, a significant, diffuse muscle weakness, and hypotonia were observed since the delivery. The primitive reflexes were poorly expressed: the sucking, rooting, palmar grasp, and plantar reflexes were partially present, while Moro reflex was absent in the patient. The weight at the time of birth was 3080 g (27th centile, − 0.62SD), the length - 56 cm (97th centile, + 1,86SD), and the head circumference - 36 cm (84th centile, + 0.98SD). After 4 months of life, the patient’s weight increased to 6420 g (57th centile, + 0.17SD), the length to 66.5 cm (96th centile, + 1,7SD), and the head circumference to 42.2 cm (77th centile, + 0.74SD), according to “PED(Z) pediatric calculator” online [13].

XLMTM patient with severe hypotonia

Facial appearance of XLMTM patient

High-arched palate and V-shaped lip

Pectus excavatum of XLMTM patient
At the beginning of the hospitalization, the child required mechanical ventilation. Then, the non-invasive CPAP (continuous positive airway pressure) support was sufficient, initially in the day and night, and later only during the sleep. After 54 days of respiratory support, at the end of the hospital stay, the infant has been breathing without any help. He was discharged home, with a pulse oximetry monitoring, oxygen support on demand (an oxygen concentrator), and a suction device. After 2 weeks, the mother observed a deterioration of the respiratory function, which resulted in rehospitalization. The child required mechanical ventilation again. After performing tracheostomy, he was discharged home again; however, mechanical ventilation had to be continued.
The patient could not swallow nutrition and saliva, but the sucking reflex was partially retained. He was fed via a nasogastric tube, and then via PEG (percutaneous endoscopic gastrostomy) tube. Before PEG placement, the gastroesophageal reflux was excluded with esophageal pH-monitoring.
In the course of the hospitalization, there was no improvement in the primitive reflexes and the muscular strength. The tendon reflexes were absent, as well. Tachycardia, tachypnea, and dyspnea were observed, when the child was crying. The volume of the voice was low. The ophthalmic consultation showed no significant abnormality in vision. The infant was able to keep the eye contact with the surroundings and to react appropriately to the sounds. What is more, he reacted well to his mother’s presence. In the USG (ultrasonography) imaging, there were no testicles present in the scrotum.
The newborn underwent a thorough diagnostic process on account of muscle weakness. The brain ultrasound at the beginning of the hospitalization was normal, but after few weeks, a slight ventriculomegaly, without increased intracranial pressure, was observed. MRI (magnetic resonance imaging) in T2-weighted image showed an increased signal in a cortical white matter (Fig. 5). The serum creatine phosphokinase level was normal, the same as the levels of ammonia, VLCFA (very-long chain fatty acids), and lactic acid. The level of 2-ketoglutaric acid in urine was elevated in organic acids profile according to GC/MS (gas chromatography/mass spectrometry) method, but the results did not indicate any congenital, metabolic defects that could have been related to them. There was also no metabolic acidosis. Therefore, the metabolic diseases, which might have been the cause of the muscle weakness, have been ruled out. As a next step, genetic analyses have been performed, as given below.

MRI image of our patient showing increased signal in cortical white matter
The child was first diagnosed for spinal muscular atrophy and Prader-Willi syndrome. The first MLPA (multiplex ligation-dependent probe amplification) analysis (SALSA MLPA P060 SMA kit) showed the presence of two copies of exon 7 and 8 of SMN1 gene and one copy of exon 7 and 8 of SMN2 gene (rsa SMN1ex7(P060)x2, rsa SMN1ex8(P060)x2, rsa SMN2ex7(P060)x1, rsa SMN2ex8(P060)x1). The second MLPA analysis (SALSA MLPA ME028 Prader Willi/Angelman kit) showed no deletion in the region 15q11 (rsa (ME028)x2) and proved the correct methylation pattern in this locus. These two analysis excluded the abovementioned diseases.
As a next step, the arrayCGH (array comparative genomic hybridization) analysis was performed (Agilent ISCA 8x60K V2), with normal results, what excluded the presence of pathological CNVs (copy number variations) in this patient.
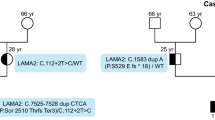
Subsequently, the NGS-based panel analysis was performed. The peripheral blood sample of the patient was sent to Blueprint Genetics Company’s Laboratory in Helsinki, where the DNA sequence and copy number variation analyses were performed (The Blueprints Genetics Comprehensive Muscular Dystrophy/Myopathy Panel (version 4, May 01 2018) Plus Analysis). The test covered 73 genes, proven to be connected with congenital myopathy. The results have shown that the patient is hemizygous for MTM1 mutation c.197C>G, p.(Thr66Arg), which is a variant of uncertain significance (VUS). The variant was classified according to Blueprint Genetics Variant Classification Schemes modified from ACMG guideline 2015 [14]. The laboratory performed also further bioinformatical tests. In silico analysis was executed with Polyphen, MutationTaster, and SIFT software. All tools showed a high probability of the variant being damaging and thus a possible connection between the mutation and the proband’s phenotype. The presence of this variant has been confirmed in the patient’s second blood sample with targeted Sanger sequencing. Then, the family history has been analyzed and further tests were performed: patient’s mother, father, healthy brother, and mother’s twin brother have been tested with the targeted Sanger sequencing for this variant. Mother turned out to be a heterozygous carrier of this variant, whereas the mutation was absent in the father, healthy brother, and mother’s twin brother (Fig. 6). Segregation analysis, with the absence of the variant in healthy males, strengthens the probability that the variant is causative in presented patient.

Pedigree of our patient’s family
Discussion
The patient that we are reporting on was diagnosed with a c.197C>G, p.(Thr66Arg) MTM1 mutation, described as a variant of uncertain significance, as there is currently insufficient proof for this mutation to be pathogenic. To date, it has not been described in the medical literature or in genome databases. However, the laboratory in silico analyses showed a high probability of the variant being deleterious.
According to the medical literature, VUS is not a common finding in patients with XLMTM phenotype. In the recently carried out multicenter, retrospective medical record review [8], only one patient out of 106 was diagnosed with VUS mutation (a single nucleotide intron 4 variant predicted to generate a novel splice acceptor site 16 bases proximal to the exon 5 splice acceptor). Also in the same study, most of the mutations responsible for the symptoms were described as familial, based on either positive family history or maternal genetic testing [8]. According to Dowling et al. [6], only 10–20% of patients carry de novo mutation. In our patient’s DNA, we found a familial mutation, as the genetic test proved the mother to be a female carrier.
Patient’s phenotype correlates strongly with the hitherto descriptions of the MTM1 gene’s mutations. According to Dowling et al. [6], 80% of patients with XLMTM present with classic, severe form of the disease, with significant hypotonia, respiratory failure, and weakness, and these findings are consistent with the broad spectrum of symptoms in our patient. Other characteristic neuromuscular findings, that our patient had, include areflexia, diminished muscle bulk, and delayed motor milestones. Interestingly, he did not present any ophthalmoplegia or ptosis.
Gestational history has also been of interest. Although polyhydramnios and decreased fetal movements were reported in respectively 50–57% and 37–50% of patients with XLMTM, and about one third of them was born prematurely [8, 15], this was not the case in our proband.
Typical XLMTM dysmorphological changes consist of dolichocephaly with midface retrusion, high-arched palate, disproportionately large body length and head circumference, in comparison to lower weight, which our patient presented, and also long fingers and toes, which were absent in our patient [12]. Additional findings described in medical literature are hepatic peliosis, cardiac arrythmias, gastroesophageal reflux, gallstones, kidney stones, clubfeet, hip dysplasia, and cryptorchidism, yet our patient suffered only from the last one [6]. It is not ruled out, that some of the other abovementioned disorders will show up in the future.
In many publications, an important element of X-linked myotubular myopathy’s diagnosis is the muscle biopsy. In the study by Beggs et al. [8], 80% of participants were tested in this way, while nearly all of them (96%) had the diagnosis confirmed with genetic testing. Some authors consider muscle biopsy an essential element of the diagnosis, although it is not pathognomonic [16]. Conversely, Dowling et al. in their comprehensive study on XLMTM [6] put emphasis rather on clinical findings and genetic testing, than on histopathological changes. Taking under consideration the well-being of our patient, the invasiveness of biopsy, and our opinion, that genetic analysis outcomes should be decisive in making a diagnosis, the patient did not have the histopathological examination done. In our opinion, it was not necessary, concerning his clinical phenotype and the conclusions of genetic testing.
The prognosis for children with XLMTM is still highly unfavorable. Approximately 25% of male neonates die in the first year of life [6]. In a recent cross-sectional and prospective investigations [15] with a median patients’ age 8 years 4 months, 77% of participants required respiratory, feeding, and wheelchair support, while 86% of them at least one form of the abovementioned aids. The mortality was estimated for 10% per year, nearly in all cases because of the respiratory failure. Apart from significant muscle weakness, patients suffered from many other health issues, as for example severe scoliosis, gastrointestinal reflux, and bone fractures. What is more, the course of the disease was either slowly progressive or stationary. In 1 year of longitudinal observation, 73% of patients noted no changes or small changes in respiratory support requirement, whereas requirements of 18% increased. Currently, there are no medications that could modify the course of the disease. The treatment focuses on respiratory and nutritional support, together with prevention of various complications and accompanying diseases. Fortunately, the gene therapy for XLMTM is currently under investigation. A Gene Transfer Clinical Study in X-Linked Myotubular Myopathy started in 2017 [17] and raises hope for the future effective treatment of XLMTM patients.
Conclusion
We report a case of a male infant with a mutation of MTM1 gene and centronuclear myopathy phenotype. Although the genetic variant is described as one of uncertain significance, the spectrum of symptoms and the distribution of mutation among patient’s relatives let us suspect that it may be with high probability pathogenic. In this way, we hope that our finding could contribute to the diagnostic process of subsequent patients.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Abbreviations
- XLMTM:
-
X-linked myotubular myopathy
- HGMD:
-
Human Gene Mutation Database
- VUS:
-
Variant of uncertain significance
- CPAP:
-
Continuous positive airway pressure
- PEG:
-
Percutaneous endoscopic gastrostomy
- USG:
-
Ultrasonography
- MRI:
-
Magnetic resonance imaging
- VLCFA:
-
Very-long chain fatty acids
- GC/MS:
-
Gas chromatography/mass spectrometry
- MLPA:
-
Multiplex ligation-dependent probe amplification
- arrayCGH:
-
Array comparative genomic hybridization
- CNVs:
-
Copy number variations
References
Hohendahl A, Roux A, Galli V (2016) Structural insights into the centronuclear myopathy-associated functions of BIN1 and dynamin 2. J Struct Biol 196(1):37–47
OMIM - Online Mendelian Inheritance in Man Database. https://www.omim.org/entry/300415 Accessed 9 Jan 2021.
Al-Qusairi L, Weiss N, Toussaint A, Berbey C, Messaddeq N, Kretz C et al (2009 Nov 3) T-tubule disorganization and defective excitation-contraction coupling in muscle fibers lacking myotubularin lipid phosphatase. Proc Natl Acad Sci U S A 106(44):18763–18768
Dowling JJ, Vreede AP, Low SE, Gibbs EM, Kuwada JY, Bonnemann CG, et al. Loss of myotubularin function results in T-tubule disorganization in zebrafish and human myotubular myopathy. Cox GA, editor. PLoS Genet. 2009;5(2):e1000372.
Laporte J, Blondeau F, Buj-Bello A, Mandel JL (2001) The myotubularin family: from genetic disease to phosphoinositide metabolism. Trends Genet 17(4):221–228
Dowling J, Lawlor M, Das S (2002) X-linked myotubular myopathy. GeneReviews® 300415:1–24
Herman GE, Finegold M, Zhao W, de Gouyon B, Metzenberg A (1999) Medical complications in long-term survivors with X-linked myotubular myopathy. J Pediatr 134(2):206–214
Beggs AH, Byrne BJ, De Chastonay S, Haselkorn T, Hughes I, James ES et al (2018) A multicenter, retrospective medical record review of X-linked myotubular myopathy: the RECENSUS study. Muscle Nerve 57:550–560
Laporte J, Biancalana V, Tanner SM, Kress W, Schneider V, Wallgren-Pettersson C et al (2000) MTM1 mutations in X-linked myotubular myopathy. Hum Mutat 15(5):393–409
Herman GE, Kopacz K, Zhao W, Mills PL, Metzenberg A, Das S (2002) Characterization of mutations in fifty North American patients with X-linked myotubular myopathy. Hum Mutat 19(2):114–121
Biancalana V, Caron O, Gallati S, Baas F, Kress W, Novelli G et al (2003) Characterisation of mutations in 77 patients with X-linked myotubular myopathy, including a family with a very mild phenotype. Hum Genet 112(2):135–142
Tsai TC, Horinouchi H, Noguchi S, Minami N, Murayama K, Hayashi YK et al (2005) Characterization of MTM1 mutations in 31 Japanese families with myotubular myopathy, including a patient carrying 240 kb deletion in Xq28 without male hypogenitalism. Neuromuscul Disord 15(3):245–252
https://www.pedz.de/en/calculators.html. 2019. p. https://www.pedz.de/en/calculators.html.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–423
Amburgey K, Tsuchiya E, De Chastonay S, Glueck M, Alverez R, Nguyen CT et al (2017) A natural history study of X-linked myotubular myopathy. Neurology. 89(13):1355–1364
Jungbluth H, Wallgren-Pettersson C, Laporte J (2008) Centronuclear (myotubular) myopathy. Orphanet J Rare Dis 3:26
Gene Transfer Clinical Study in X-Linked Myotubular Myopathy - Full Text View - ClinicalTrials.gov [Internet]. [cited 2019 Jun 30]. Available from: https://clinicaltrials.gov/ct2/show/study/NCT03199469?cond=X+Linked+Myotubular+Myopathy&rank=2
Acknowledgements
Not applicable
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
PK, KS, KK, TT, AG, MJ, and WD made substantial contribution to the conception of the patient’s diagnostic process, conducted the diagnostic procedures, and collected and interpreted clinical data concerning the patient. AD, WN, KK, and TT were major contributors in drafting the initial version of the manuscript. PK reviewed and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Written informed consent was obtained from patient’s parents for publication of this case report and accompanying images.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dudzik, A., Nedza, W., Końska, K. et al. A novel mutation in MTM1 gene in newborn, resulting in centronuclear myopathy phenotype: a case report. Egypt J Med Hum Genet 22, 19 (2021). https://doi.org/10.1186/s43042-021-00140-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-021-00140-5