
Abstract
Background
Tetralogy of Fallot is the most prevalent cyanotic congenital heart disease, occurring in 1/3 600 live births. This disorder comprises ventricular septal defect, right ventricular outflow obstruction, over-riding aorta, and right ventricular hypertrophy. The present study aims to reveal the spectrum of Nk2 homeobox 5 (NKX2-5) variants identified in a Moroccan non-syndromic tetralogy of Fallot cohort and to compare mutation rate with different studies from all over the world. Thirty-one patients with non-syndromic tetralogy of Fallot were recruited in this cross-sectional study. DNAs were extracted, and coding regions of NKX2.5 were PCR-amplified and sequenced. The obtained sequences were analyzed using different bioinformatics tools. Statistical comparisons were carried out using the R software.
Results
R25C mutation was found in two patients, in association with the E21E variant. The latter variant was frequently observed in the population and seems to have a potential altering effect on the splicing process. The NKX2.5 mutation rate in our tetralogy of Fallot population is around 6.4%, and no significant difference was noticed in comparison with previous studies. At the same time, a comparison of R25C mutation rate between atrial septal defect and tetralogy of Fallot worldwide populations shows a particular association of R25C mutation with tetralogy of Fallot phenotype.
Conclusions
This study reveals a consistency between our NKX2.5 mutation rate and those of different tetralogy of Fallot populations around the world. Our findings suggest a possible combined effect of R25C mutation and E21E variant on the carriers and emphasize particularly the significant association of R25C mutation with tetralogy of Fallot, which highlights the importance of an anticipative screening for TOF phenotype among the carriers’ offspring at the perinatal period.
Similar content being viewed by others

Background
Congenital heart disease (CHD) is the most common form of birth anomalies, with a prevalence of 1% of newborns [1]. This congenital disorder results from both genetic and environmental etiologies and is considered the first non-infectious cause of death in the first year of life [2, 3]. Tetralogy of Fallot (TOF) constitutes the most prevalent cyanotic congenital heart disease, occurring in 1/3 of 600 live births and accounting for 6.8% of congenital heart diseases [4, 5]. This right-to-left shunt disorder comprises ventricular septal defect, right ventricular outflow obstruction, over-riding aorta, and right ventricular hypertrophy.
Up to 25% of TOF cases occur in the setting of syndromic conditions, notably DiGeorge syndrome (22q11 microdeletion) [6].
Genetic etiology involved in non-syndromic tetralogy of Fallot includes most often cardiac transcription factors, such as Nk2 homeobox 5 (NKX2-5) (5q35), GATA-binding protein 4 (GATA4) (8p23.1), and T-BOX 1 (TBX1) (22q11.21). In the normal cardiogenesis process, these genes contribute particularly to the differentiation of the second heart field (SHF) and the heart looping [7,8,9,10].
NKX2-5, a member of the NKX family, is a homeobox transcription factor with a central role in regulating heart development. It interacts with GATA4 and serum response factor (SRF) throughout cardiogenesis to activate cardiac sarcomeric genes [11]. NKX2-5, mapped in 5q35, is the first genetic factor to be associated with non-syndromic congenital heart disease, particularly with non-syndromic atrial septal defect (ASD) combined with atrioventricular block [12]. Afterward, several studies confirmed the association of NKX2-5 mutations with other types of congenital heart disease, notably tetralogy of Fallot and ventricular septal defect [8, 13, 14].
In the present study, we carried out a mutational screening of NKX2-5 in a Moroccan cohort with tetralogy of Fallot to reveal the potential pathogenic mutations that may cause such phenotype. In the second part, we compared our findings with those reported in different populations. The third part of this paper was focused on assessing the R25C variant prevalence among worldwide populations affected by either tetralogy of Fallot or atrial septal defect (ASD).
Methods
Patients
The thirty-six unrelated patients, recruited in this cross-sectional study, were initially evaluated by the Medico-surgical Unit of the Cardio-pediatric Department. After confirming the diagnosis of tetralogy of Fallot by electrocardiography (ECG) and Doppler echocardiography, the patients underwent clinical evaluation, karyotyping, and 22q11 microdeletion testing using the fluorescent in situ hybridization (FISH) technique, in the Medical Genetics Laboratory. Patients with tetralogy of Fallot phenotype, normal karyotype, and no 22q11 microdeletion were included in the study. Five patients, in whom a syndromic association was confirmed or suspected, were excluded from the study. All included patients were interviewed, and the history of personal and family disorders was noted. This work was approved by the local ethics committee, under reference number 06/14.
Molecular analysis
After obtaining written informed consent, the peripheral blood was obtained from the 31 patients with non-syndromic tetralogy of Fallot, and DNA samples were extracted from blood lymphocytes using the optimized salting-out technique [15].
We carried out polymerase chain reaction (PCR) to amplify NKX2-5 coding exons and their flanking introns using the primer pairs derived from previously published data [16]. Primer sequences with PCR product sizes are illustrated in Table 1.
PCR reaction mix was prepared in a 25-mL final volume containing 10 pmol of each primer, 1× PCR buffer (Invitrogen, CA, USA), 15–25 mM MgCl2, 10 mM dNTP, 1 U of Taq (Invitrogen, CA, USA), and 40 ng of genomic DNA. The PCR cycling conditions performed in the Veriti 96-well Thermal Cycler 9902 (Applied Biosystems, MA, USA) were 94 °C for 7 min; 35 cycles of 94 °C for 40 s, 59–64 °C (according to the specific hybridization temperature of each primer pairs) for 30 s, and 72 °C for 40 s; and 72 °C for 7 min.
After electrophoresis, purified PCR product underwent direct sequencing using the BigDye Terminator V1.1 Cycle Sequencing Kit (ABI Prism, MA, USA) and the Applied Biosystems 3500Dx Genetic Analyser.
In silico analysis
Chromatograms were analyzed by the Sequencing Analysis SeqA v.5.4 (Applied Biosystems, MA, USA). The obtained sequences were analyzed using various bioinformatics analysis tool, notably “Nucleotide Blast” for pairwise alignment (NCBI); Mutation Taster2, SIFT v5.1.1, SNAP2, and SNPs&GO to predict the functional impact of non-synonymous variants; and Human Splicing Finder (HSF 3.0) to assess the impact of silent variants on splicing process.
Statistical analysis
The statistical tests used to compare the mutational rate between study cohorts were Fisher’s exact test for reduced cohort size and chi-square tests in larger cohort size. Tests were performed using the R software.
Results
Thirty-one patients suffering from tetralogy of Fallot were screened for NKX2.5 mutations. The studied population was composed of 17 males and 14 females with a sex ratio of 1.2. The mean age at enrolment was 6 years old, with children ages ranging from 2 months to 15 years. About 21% of the patients were born to consanguineous parents. Maternal and paternal mean ages at patients’ birth were 28 and 38 years, respectively.
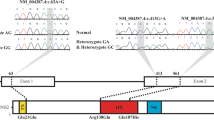
Sanger sequencing of coding regions showed two variants in exon 1. The first substitution (c.63A>G) is a synonymous recurrent variant, being detected in 61% of the studied cohort. The second substitution (c.73C>T) is a missense variant that was found in 6.45% of the affected group. Table 2 and Fig. 1 show more details about these variants.

Sequencing profiles of detected variants (a, b) and organization of NKX2.5 domains with detected variants location (c)
Functional prediction tools show a pathogenic impact of the R25C (c.73C>T) variant. Mutation taster2 algorithm predicts a disease-causing effect with a score of 180, Sift algorithm concludes a damaging effect with a score of 0.05, SNAP2 algorithm shows an altering effect with a score of 76, while SNPs&GO algorithm considers R25C substitution as a disease-related variant. On the other hand, HSF algorithms show a potential altering effect of the E21E variant on splicing activity, as illustrated in Fig. 2.

HSF output for pathogenic effect prediction of the E21E variant
Considering only the missense variants, the mutation rate in our cohort seems to be around 6.45%. This rate was compared with NKX2.5 mutation rates of various populations affected by tetralogy of Fallot from all over the world. Table 3 shows the p values of different comparisons.
Discussion
NKX2-5 protein is one of the most important transcription factors in heart development during the embryogenesis stage. It is notably involved in regulating cardiomyocyte differentiation and proliferation, through different stages of cardiogenesis. During heart formation, NKX2.5 protein plays a central role in the second heart field (SHF) differentiation and the heart looping process. These heart areas constitute particularly the major parts damaged in the case of tetralogy of Fallot, which suggests a strong involvement of NKX2.5 mutations in this particular congenital heart disease. For this reason, in the present work, the NKX2.5 gene was chosen to be screened in a group of thirty-one patients with non-syndromic tetralogy of Fallot. This cohort has been screened for GATA4 mutations in an earlier paper [25], but no confirmed pathogenic mutation was revealed. Clinical and environmental investigation findings were discussed in that study and have shown a strong association between TOF occurrence and maternal passive smoking.
We detected two variants, localized both in the first exon of NKX2.5. The first substitution, c.73C>T (rs28936670), is a heterozygous missense variant that replaces arginine amino acid with a cytosine at position 25 of the NKX2.5 coding sequence. This variant was observed in two female patients, one of them is issued from a consanguineous marriage.
The second substitution, c.63A>G (rs2277923), is a synonymous variant that does not change the corresponding amino acid, which is glutamate, at position 21 of the coding sequence. rs2277923 is a recurrent variant that was found in 19 patients as heterozygous and homozygous substitutions, respectively in 73.7% and 26.3% of carriers.
Both of the detected variants are very close to the tinman domain (TN). The latter has a pivotal role in activating the transcription of NKX2.5 downstream-targeted genes [26].
In silico analysis of R25C mutation pathogenic impact shows a damaging and disease-causing effect through four different prediction programs, namely, Sift, Mutation taster2, SNAP2, and SNPs&GO algorithms. Given that R25 residue is a relatively conserved amino acid, this substitution could be considered as a probably pathogenic variant.
R25C variant was proved, through previous independent functional studies, to impair dimer formation at the dimeric binding sites of target genes and to significantly decrease transactivation of downstream gene promoters [27, 28].
On the other hand, HSF algorithms that assess the effect of silent variants on splicing machinery show an interesting potential altering impact for the E21E variant, by creating an exonic splicing silencer (ESS) site and altering an exonic splicing enhancer (ESE) site (Fig. 2). These effects could disrupt the splicing process by splicing out the entire concerned exon, which may lead to a truncated protein most probably inactive. This conclusion should be obviously confirmed by additional in vitro functional studies.
It is worth noting that the E21E variant (rs2277923) was detected in both carriers of the R25C mutation, as a homozygous substitution in one of them. This needs to be further studied to confirm whether such compound variants have a more pathogenic impact on the NKX2.5 protein function.
Therefore, by considering only missense variants, the NKX2.5 mutation rate in our study seems to be around 6.4%. In the second part of this study, we compared this rate to those obtained in different previous studies. The results in Table 3 show an absence of significant differences between our rate and rates found in diverse TOF populations throughout the world. Moreover, no significant differences were found when comparing these study rates with each other.
R25C mutation is considered the most recurrent NKX2.5 variant in CHD patients [7, 12, 19]. This hotspot variant was detected worldwide in various CHD subtypes, including TOF, ASD, ventricular septal defect (VSD), hypoplastic left heart (HLHS), interrupted aortic arch (IAA), and atrioventricular connection (AVC) [29]. In the third part of this study, we compared the R25C mutation cumulative frequency between TOF and ASD populations deducted from 18 studies from all over the world. The corresponding results in Table 4 show a significant difference, p = 0.015, meaning that R25C mutation is significantly associated with TOF phenotype compared to ASD. This could suggest that, in the case of R25C mutation occurrence, there may be a relatively higher likelihood that the embryo develops a TOF phenotype rather than ASD.
This hypothesis seems to be supported by previous studies, in which the authors emphasized the significantly higher recurrence of this variant among TOF patients [7, 18], with a potential increased risk of TOF manifestation among R25C mutation carriers children [37].
These findings would particularly encourage anticipative screening for TOF phenotype among offspring of R25C mutation carriers, especially at the perinatal period. Given the life-threatening complications related to this disorder at the perinatal period [38], such an anticipative step may constitute an additional support to allow providing suitable care for the newborn, starting from his/her earliest stages of life.
Conclusions
In conclusion, the present study reports, for the first time, NKX2.5 variants found in a Moroccan population affected by tetralogy of Fallot. NKX2.5 mutation rate determined in our study population (6.4%) is consistent with international rates. Our findings suggest a potential impact of E21E variant on splicing process and a possible combined effect of both R25C mutation and E21E variant on our mutated patients and emphasize particularly the significant association of R25C mutation with tetralogy of Fallot, which may encourage the anticipative screening for TOF phenotype among R25C mutation carriers’ offspring at the perinatal period.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ASD:
-
Atrial septal defect
- AVC:
-
Atrioventricular connection
- CHD:
-
Congenital heart disease
- ECG:
-
Electrocardiography
- ESE:
-
Exonic splicing enhancer
- ESS:
-
Exonic splicing silencer
- FISH:
-
Fluorescent in situ hybridization
- GATA4:
-
GATA-binding protein 4
- HLHS:
-
Hypoplastic left heart
- HSF:
-
Human Splicing Finder
- IAA:
-
Interrupted aortic arch
- NKX2-5:
-
Nk2 homeobox 5
- PCR:
-
Polymerase chain reaction
- SHF:
-
Second heart field
- SRF:
-
Serum response factor
- TBX1:
-
T-BOX 1
- TN:
-
Tinman
- TOF:
-
Tetralogy of Fallot
- VSD:
-
Ventricular septal defect
References
Barnett P, den Boogaard M van, Christoffels V. Localized and temporal gene regulation in heart development. Vol. 100, Current Topics in Developmental Biology. Elsevier Inc.; 2012. 171–201 p.
Hoffman JIE, Kaplan S (2002) The incidence of congenital heart disease. J Am Coll Cardiol. 39(12):1890–1900
Garg V (2006) Insights into the genetic basis of congenital heart disease. Cell Mol Life Sci C. 63(10):1141–1148
Shinebourne E, Anderson R. Fallot’s tetralogy. In: Anderson R, Baker E, Macartney F, Rigby M, Shinebourne E, Tynan M, editors. Paediatric cardiology. 2nd ed. Toronto: Churchill Livingstone, London; 2002. p. 1213–502.
Ferencz C, Rubin J, McCarter R, BRENNER J, Neill C, Perry L et al (1985) Congenital heart disease: prevalence at livebirth: the Baltimore- Washington Infant Study. Am J Epidemiol. 121:31–36
Davis S (2006) Tetralogy of Fallot with and without pulmonary atresia. In: Nichols DG, Ungerleider RM, Spevall P (eds) Critical Heart Disease in Infants and Children, 2nd edn. Mosby, Philadelphia, PA, pp 755–766
Harvey R, Lai D, Elliott D, Biben C, Solloway M, Prall O et al (2002) Homeodomain factor Nkx2-5 in heart development and disease. Cold Spring Harb Symp Quant Biol. 67:107–114
Goldmuntz E, Geiger E, Benson DW (2001) NKX2.5 mutations in patients with tetralogy of Fallot. Circulation. 104(21):2565–2568
Peterkin T, Gibson A, Loose M, Patient R (2005) The roles of GATA-4, -5 and -6 in vertebrate heart development. Semin Cell Dev Biol. 16:83–94
Jerome L, Papaioannou V (2001) DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat Genet. 27:286–291
Sepulveda J, Vlahopoulos S, Iyer D, Belaguli N, Schwartz R (2002) Combinatorial expression of GATA4, Nkx2-5, and serum response factor directs early cardiac gene activity. J Biol Chem. 277:25775–25782
Schott JJ, Benson DW, Basson CT, Pease W, Silberbach GM, Moak JP, et al. Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science (80-). 1998;281(5373):108–11.
Benson DW, Silberbach GM, Kavanaugh-McHugh A, Cottrill C, Zhang Y, Riggs S et al (1999) Mutations in the cardiac transcription factor NKX2.5 affect diverse cardiac developmental pathways. J Clin Invest. 104(11):1567–1573
McElhinney DB, Geiger E, Blinder J, Benson DW, Goldmuntz E. NKX2.5 mutations in patients with congenital heart disease. 2003;42(9):391–5.
Miller SA, Dykes D, Polesky H (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 16:12–15
Wang J, Xin YF, Liu XY, Liu ZM, Wang XZ, Yang YQ (2011) A novel NKX2-5 mutation in familial ventricular septal defect. Int J Mol Med. 27(3):369–375
Hussein IR, Helmy NA, Hussein HA, Bassyouni R, Ahmed A (2009) Genetic studies of congenital heart defects in Egyptian patients. Medicine (Baltimore). 4(1):55–66
Hussein IR, El-Ruby MO, Fahmi AA, El-Desouky MA, Fayez AE-DG (2012) Detection of mutations in GATA4 and Nkx2.5 genes in patients with Fallot’s tetralogy. Middle East J Med Genet. 1(1):49–52
Akçaboy MI, Cengiz FB, Inceoǧlu B, Uçar T, Atalay S, Tutar E et al (2008) The effect of p.Arg25Cys alteration in NKX2-5 on conotruncal heart anomalies: mutation or polymorphism? Pediatr Cardiol. 29(1):126–129
Rauch R, Hofbeck M, Zweier C, Koch A, Zink S, Trautmann U et al (2010) Comprehensive genotype-phenotype analysis in 230 patients with tetralogy of Fallot. J Med Genet. 47(5):321–331
Gioli-Pereira L, Pereira AC, Mesquita SM, Xavier-Neto J, Lopes AA, Krieger JE (2010) NKX2.5 mutations in patients with non-syndromic congenital heart disease. Int J Cardiol. 138(3):261–265
Wang J, Liu XY, Yang YQ (2011) Novel NKX2-5 mutations responsible for congenital heart disease. Genet Mol Res. 10(4):2905–2915
Zhang W, Li X, Shen A, Jiao W, Guan X, Li Z (2009) Screening NKX2.5 mutation in a sample of 230 Han Chinese children with congenital heart diseases. Genet Test Mol Biomarkers. 13(2):159–162
Kodo K, Nishizawa T, Furutani M, Arai S, Ishihara K, Oda M et al (2012) Genetic analysis of essential cardiac transcription factors in 256 patients with non-syndromic congenital heart defects. Circ J. 76(7):1703–1711
EL Bouchikhi I, Belhassan K, Moufid FZ, Houssaini MI, Laila B, Imane S, et al. GATA4 molecular screening and assessment of environmental risk factors in a Moroccan cohort with tetralogy of Fallot. 2018;18(4):922–30.
Pradhan L, Genis C, Scone P, Weinberg E, Kasahara H, Nam H (2012) Crystal structure of the human NKX2.5 homeodomain in complex with DNA target. Biochemistry. 51(32):6312–6319
Kasahara H, Lee B, Schott JJ, Benson DW, Seidman JG, Seidman CE et al (2000) Loss of function and inhibitory effects of human CSX/NKX2.5 homeoprotein mutations associated with congenital heart disease. J Clin Invest. 106(2):299–308
Dentice M, Cordeddu V, Rosica A, Ferrara AM, Santarpia L, Salvatore D et al (2006) Missense mutation in the transcription factor NKX2-5: a novel molecular event in the pathogenesis of thyroid dysgenesis. J Clin Endocrinol Metab. 91(4):1428–1433
Kalayinia S, Ghasemi S, Mahdieh N. A comprehensive in silico analysis, distribution and frequency of human Nkx2-5 mutations; a critical gene in congenital heart disease. J Cardiovasc Thorac Res [Internet]. 2019;11(4):287–299. Available from: https://doi.org/https://doi.org/10.15171/jcvtr.2019.47
Elliott DA, Kirk EP, Yeoh T, Chandar S, McKenzie F, Taylor P et al (2003) Cardiac homeobox gene NKX2-5 mutations and congenital heart disease: associations with atrial septal defect and hypoplastic left heart syndrome. J Am Coll Cardiol. 41(11):2072–2076
Cao Y, Wang J, Wei C, Hou Z, Li Y, Zou H et al (2016) Genetic variations of NKX2-5 in sporadic atrial septal defect and ventricular septal defect in Chinese Yunnan population. Gene. 575(1):29–33
Liu XY, Wang J, Yang YQ, Zhang YY, Chen XZ, Zhang W et al (2011) Novel NKX2-5 mutations in patients with familial atrial septal defects. Pediatr Cardiol. 32(2):193–201
Stallmeyer B, Fenge H, Nowak-Göttl U, Schulze-Bahr E (2010) Mutational spectrum in the cardiac transcription factor gene NKX2.5 (CSX) associated with congenital heart disease. Clin Genet. 78(6):533–540
Sarkozy A, Conti E, Neri C, D’Agostino R, Digilio MC, Esposito G et al (2005) Spectrum of atrial septal defects associated with mutations of NKX2.5 and GATA4 transcription factors. J Med Genet. 42(2):2–7
Hirayama-Yamada K, Kamisago M, Akimoto K, Aotsuka H, Nakamura Y, Tomita H, et al. Phenotypes with GATA4 or NKX2.5 mutations in familial atrial septal defect. Am J Med Genet. 2005;135 A(1):47–52.
Abou Hassan OK, Fahed AC, Batrawi M, Arabi M, Refaat MM, Depalma SR et al (2015) NKX2-5 mutations in an inbred consanguineous population: genetic and phenotypic diversity. Sci Rep. 5:1–8
Morgenthau A, Frishman WH (2018) Genetic origins of tetralogy of Fallot. Cardiol Rev. 26(2):86–92
Forman J. A review of tetralogy of Fallot and postoperative management. Crit Care Nurs Clin NA [Internet]. 2019;31(3):315–28. Available from: https://doi.org/https://doi.org/10.1016/j.cnc.2019.05.003
Acknowledgements
We thank the patients and their families for their participation in this study.
Funding
This work was financially supported by the Hassan II University Hospital and Faculty of Medicine and Pharmacy of Fez, University of Sidi Mohamed Ben Abdellah, Morocco. The funders had no role in any aspect of the study (neither in the design of the study, collection, analysis, and interpretation of data nor in writing the manuscript).
Author information
Authors and Affiliations
Contributions
Conception and supervision of study: IEB, MIH, KO, and SA. Research techniques: IEB and FZM. Analysis and interpretation of the data: IEB and MIH. Writing of the paper: IEB. Critical review: FZM. Clinical assessment: KB, LB, IS, and SA. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the local ethic committee entitled “Comité d’éthique de la Faculté de Médecine et de Pharmacie et du CHU Hassan II de Fès” (located in Faculty of Medicine and Pharmacy of Fez) under the reference ID: Ref. 06/14. Written informed consent was obtained from participating patients or their tutors.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
EL Bouchikhi, I., Belhassan, K., Moufid, F.Z. et al. Screening of NKX2.5 gene in Moroccan Tetralogy of Fallot (TOF) patients: worldwide mutation rate comparisons show a significant association between R25C variant and TOF phenotype. Egypt J Med Hum Genet 22, 18 (2021). https://doi.org/10.1186/s43042-021-00136-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-021-00136-1