
Abstract
Background
Pitt-Hopkins syndrome (PHS) is a rare cause of severe intellectual disability, seizures, language impairment, and peculiar facial dysmorphism. It is caused by a mutation in transcription factor 4 (TCF4). Through molecular karyotyping and mutational analysis, a study identified recessive defects in two genes, contactin associated protein like 2 (CNTNAP2) and Neurexin I (NRXN1), in patients with similar presentations of Pitt-Hopkins syndrome and called Pitt-Hopkins-like syndrome (Zweier et al., J Med Genet 80: 994-1001, 2007).
We present the first case report of a child in Iraq with Pitt-Hopkins-like syndrome that was referred to the Welfare Children’s Hospital/Medical City of Baghdad because of her intellectual disability.
Case presentation
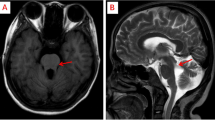
The patient was 4-year-old female child who presented with psychomotor delay and language impairment. She had frequent attacks of the respiratory tract and eye infections. Ophthalmologic examination revealed left-sided esotropia and severe myopia. Routine hematologic, serologic, and chemistry tests were within normal ranges. EEG revealed diffuse theta slowing and diffuse beta activity. The audiological test was normal. NCS and EMG showed normal results. Echo study, chest X-ray, and abdominal/pelvic ultrasound revealed normal findings. Brain MRI showed mild bilateral frontal-temporal atrophy. Whole-exome sequencing (WES) revealed a homozygous stop mutation in CNTNAP2 with a heterozygous state in both parents.
Conclusion
Intellectual disability may result from different types of abnormal cellular processes and with widening the use of molecular gene analysis in cases of intellectual disability, underdiagnosed cases of Pitt-Hopkins and Pitt-Hopkins-like syndromes may be uncovered.
Similar content being viewed by others


Background
Pitt-Hopkins syndrome (PHS) is a rare cause of severe intellectual disability first detected in 1978 in two patients [1]. The syndrome is characterized by intellectual dysfunction, seizures, language impairment, postnatal growth retardation, and microcephaly. This syndrome is characterized by a particular breathing pattern that appears in mid-childhood and manifests as paroxysms of dyspnea followed by apnea and occasional cyanosis. This breathing anomaly shows no relation to epilepsy [2,3,4]. It is characterized by abnormal facial features including beaked nose with flared nostrils, deep-set eyes, fleshy lips with a typical cupid-bow upper lip, protruding lower face, and cup-shaped ears [5]. Other signs include happy demeanor, muscular hypotonia, arm flapping, hand biting, severe constipation, hypoplastic corpus callosum, and bulging caudate nuclei on brain magnetic resonance imaging (MRI) [6]. Other clinical manifestations include scoliosis, gastroesophageal reflux [7], pyloric stenosis [5], and constipation. Hearing defects have been reported in 10% of cases. Strabismus and myopia (severe myopia) are frequent findings. Sleeping disturbances have been reported in a few cases.
Pitt-Hopkins syndrome is caused by a mutation in transcription factor 4 (TCF4) which encodes a class I basic helix-loop-helix transcription factor [3,4,5]. Patients with Pitt-Hopkins syndrome have significant phenotypic similarities with Angelman and, Rett syndromes and 1q43q44 microdeletion syndrome [4, 5]. Pitt-Hopkins syndrome is frequently misdiagnosed if not supported by genetic evidence [5].
In a multicenter genetic study, only 12% of referred patients with suspected PTHS showed mutations in TCF4 [5]. Through molecular karyotyping and mutational analysis, researchers identified recessive defects in two genes, contactin-associated protein-like 2 gene (CNTNAP2) and Neurexin I (NRXN1), in patients with very similar severe intellectual disabilities and variable additional symptoms, such as seizures and breathing anomalies, characteristic of Pitt-Hopkins syndrome. These syndromes with similar presentation to Pitt-Hopkins syndrome but with CNTNAP2 or Neuroxin mutations are therefore called Pitt-Hopkins-like syndromes.
CNTNAP2 is one of the largest genes in the human genome [8]. It encodes contactin-associated protein 2 (CASPR2), a transmembrane protein that is distantly related to the family of neurexins and regulates neuroglia contact in vertebrates and glia-glia contact in insects [9].
We presented the first reported case in Iraq with Pitt-Hopkins-like syndrome. As we noticed that the number of cases with learning disability increased in the last few years, we think that tracking the causes with detailed investigations, including molecular studies may reveal new or uncommon diseases, narrow differential diagnosis and find a way for managing and treating cases of learning disability.
Case presentation
A 4-year-old female child of consanguineous parents was born through normal vaginal delivery after a full-term pregnancy complicated by polyhydramnios. The mother was exposed to a CT scan test in the first month of her pregnancy.
The child’s body weight was 3 kg, she was 40 cm long, and her Apgar score was 8 at 5 min of birth.
The child had psychomotor delay and very frequent respiratory tract infections. She complained of recurrent attacks of eye infections and was diagnosed with lacrimal duct stenosis which occurred at the age of one and a half year. At the age of 2 1/3 years, she had her first generalized tonic clonic seizure which recurred frequently and was partially controlled with sodium valproate and clobazam. Thereafter, she acquired abnormal behavior with hyperactivity, self-injury behaviors, and spells of crying, moaning, and stereotypical hand movements. Sleep became difficult with recurrent arousals and sleep terrors, and melatonin was prescribed resulting in improvement by a decrease in the frequency of attacks. She had no gastrointestinal complaints. She did not have dressing or toileting skills.
Examination showed a happy demeanor, microcephaly (head circumference is 45 cm), wide nasal bridge, and tip, narrow forehead, cupid-bow-shaped upper lip, wide mouth, full lips, and small hands. She is hyperactive with severe speech impairment. Ophthalmologic examination revealed left-sided esotropia and severe myopia. See Fig. 1.

A baby with intellectual disability, speech delay, broad bridge nose, cupid-bow-shaped upper lip, and a happy demeanor
Laboratory studies and neuroimaging
Routine hematologic, serologic, and chemistry tests were within normal ranges. EEG revealed diffuse theta slowing and diffuse beta activity which is likely the effect of clobazam. The audiological test was normal. A nerve conduction study and EMG showed normal results. Echo study, chest X-ray, and abdominal/pelvic ultrasound revealed normal findings. Brain MRI showed mild bilateral frontal-temporal atrophy and corpus callosum and brainstem images (by sagittal section) revealed normal findings (images were unavailable at the time of collecting patient data for reporting).
Molecular genetic analysis
Whole-exome sequencing (WES) was performed using the patient and both parents’ DNA obtained from venous blood.
It revealed CNTNAP2 (NM_014141.5) homozygous variant c.G2197T (p.E733X/p.Glu733). The baby was diagnosed with autosomal recessive Pitt-Hopkins-like syndrome-1 because of presence of the pathogenic variant (c.G2197T) in the CNTNAP2 gene. The CNTNAP2 variant c.2197G>T (p.Glu733) creates a stop codon and is classified as a pathogenic variant according to the American College of Medical Genetics and Genomics (ACMG) guidelines. A heterozygous state was detected in both parents.
Method
The quality control and quantification of the isolated DNA performed on Nanodrop spectrophotometer (Thermo Scientific) and Qubit fluorometer (Thermo Scientific), respectively. The concentration and 260/280 value obtained from the quantification of the DNA were optimal for the test. The structural integrity of the DNA was confirmed by gel electrophoresis. Agincourt AMPure XP (Beckman Coulter) was used for product purification before library preparation. The Agilent Sure Selected Human All Exon V6 Library Preparation Kit was used for library preparation prior to sequencing. The standard protocol of the kit was used for the test. After library preparation, DNA samples were sequenced using the Illumina NovaSeq ® platform. As a result of sequencing a paired end 150 bp reads which accounts for 12 GB of data and 112 × coverage were obtained.
Discussion
The CNTNAP2 gene encodes the CASPR2 protein from the neurexin family. It is a neuronal transmembrane protein localized at the juxta-paranodes of myelinated axons to mediate neuroglia interactions during nervous system development. This protein also has a role in the localization process of potassium channels in myelinated differentiating axons and is involved in human cortical histogenesis. The CNTNAP2 gene is one of the largest genes in the human genome and account for nearly 1.5% of chromosome 7. It spans 2.3 Mb of genomic DNA and encodes a full-length transcript of 24 exons. The CNTNAP2-encoded CASPR2 protein has 1331 residues and consists of a signal peptide, an F5/8 discoidin/neuropilin homology domain, 4 laminin G-binding domains, 2 epidermal growth factor-like domains, a fibrinogen-like region, a transmembrane domain, and a postsynaptic density interaction domain. Losses of a single allele of a gene may exert phenotypic effects if the gene is part of either a gene network or if the protein that it encodes interacts with other proteins [10].
Several publications indicate that losing both alleles of CNTNAP2 may provoke a specific clinical disorder and may provide insight into the remarkable clinical variability of CNTNAP2 disruptions [11,12,13,14]. Patients with homozygous losses of CNTNAP2 may present with a syndrome involving severe intellectual disability, early-onset drug-resistant epilepsy with language regression, cortical dysplasia-focal epilepsy syndrome (CDFE), communication impairments, and autistic features, albeit with some variability among these phenotypes [11,12,13,14].
When both CNTNAP2 alleles have been lost, the cell cannot generate a full-length transcript. Therefore, either a truncated mRNA and consequently a truncated protein or complete absence of any CNTNAP2 transcripts should account for the clinical phenotype [10].
Patients with intragenic losses of CNTNAP2 have variable clinical presentations. Lee et al. characterized the molecular and cellular phenotypes associated with heterozygous CNTNAP2 [10] and proposed 3 hypotheses to explain the variability among patients with intragenic CNTNAP2 losses [15]. First, the carrier parent has protective alleles that were not transmitted to the affected offspring. Second, the affected offspring had additional deleterious alleles that had either arisen de novo or were inherited from the other parent but not from the heterozygous parent. Third, CNTNAP2 alleles with structural deletions may experience incomplete penetrance and variable expressivity as a functional consequence of variable levels of the mutated CNTNAP2 gene being expressed [15].
CNTNAP2 alleles with expressed intragenic deletions may interfere with the ability to bridge the intercellular space between neurons by binding to specific partners, such as CNTN, CNTN2, DLG1, and DLG4. This postulated effect of intracellular deletions of CNTNAP2, and possibly other genes encoding proteins involved in connecting neuronal cells, represents a molecular basis for the presumed neuronal hypoconnectivity in neurodevelopmental disorders [16].
Pitt-Hopkins and Pitt-Hopkins-like syndromes are rare genetic disorders, and our patient is the first reported case with a homozygous CNTNAP2 mutation in Iraq. The presence of severe intellectual disability with psychomotor delay, epilepsy, and dysmorphic features suggests the diagnosis of a genetic syndrome. Rett and Angelman syndromes were highly suspected. However, the absence of breathing abnormalities, typical hand movements (washing movements), typical EEG findings, and different clinical courses made us think of another cause with such diverse clinical manifestations.
Another possibility and highly correlated differential diagnosis was 1q43q44 microdeletion syndrome. The association of intellectual disability, microcephaly, agenesis of the corpus callosum, and epilepsy characterizes the full neurodevelopmental phenotype. Point mutations in ZBTB18 and HNRNPU were implicated etiologies. Patients with HNRNPU point mutations have early-onset seizures with fever being an important triggering factor in most cases with global developmental delay and corpus callosum agenesis. In patients with ZBTB18 mutations, intellectual disability ranges from mild to severe, with global developmental delay; however, many can speak few words. Researchers also found corpus callosum dysgenesis by neuroimaging. Our patient shares many of the phenotypic manifestations of 1q43q44 microdeletion syndrome; however, the corpus callosum was normal and a genetic microarray study failed to reveal point mutations in ZBTB18 and HNRNPU [17].
We considered the possibility of Pitt-Hopkins syndrome diagnosis as our patient showed explicit findings of severe intellectual disability, severe speech impairment, OFC < 3SD, epilepsy, eye abnormalities (myopia, strabismus, tear duct blockade), typical facial features (narrow forehead, broad nasal bridge and tip, cupid’s-bow-shaped upper lip with wide mouth and filled lips), smiling appearance, abnormal behavior (hyperactive, self-injurious, and crying/moaning spells), stereotypical nonpurposeful hand movements, and abnormal brain MRI. The patient has a total score of > 10 according to the published criteria for Pitt-Hopkins syndrome diagnosis [16, 18]. According to the first international consensus statement [16], our patient is clinically confirmed to have PHS. For genetic confirmation, we sent the patient and her parents for WES to identify TCF4 gene mutations. Instead, molecular genetic analysis revealed a homozygous stop mutation in CNTNAP2 in the child with a heterozygous variant in both parents.
Our case is not the first one with clinical manifestations of PHS who did not show mutations in the TCF4 gene. A multicenter genetic study using molecular karyotyping and mutational analysis identified recessive defects in two genes, CNTNAP2 and NRXN1, in patients with a very similar severe intellectual disorder and variable additional symptoms, such as seizures and breathing anomalies, resembling Pitt-Hopkins syndrome [5].
A study performed comparing phenotypic manifestations of Pitt-Hopkins syndrome caused by autosomal recessive mutations in the CNTNAP2 and NRXN1 genes using the Drosophila model [11] revealed that resemblance among patients who have recessive defects in CNTNAP2 or NRXN1 is high. However, not all the patients in the study with NRXN1 mutations had epilepsy in contrast to those with CNTNAP2 recessive mutations. This might be associated with the previously observed neuronal migration anomalies in patients with CNTNAP2 mutations. However, the remaining symptoms, such as severe mental retardation and autistic behavior might be caused by overlapping synaptic anomalies.
Although the TCF4 gene mutation was originally implicated as a cause of Pitt-Hopkins syndrome with phenotypic resemblance to those having recessive CNTNAP2 and NRXN1 mutations with regard to facial aspects, the severity of mental retardation and breathing anomalies. Nevertheless, phenotypical differences were noticeable. In contrast to patients with Pitt-Hopkins syndrome, who present an equally severe delay or lack of motor and speech development, patients with CNTNAP2 or NRXN1 defects, also had severely impaired in speech development, present with normal or only mild to moderate delay in motor milestones. These findings are in line with the previously reported specific involvement of CNTNAP2 in language development.
Conclusion
In conclusion, intellectual disability may result from different types of abnormal cellular processes and the constellation of clinical features together with distinct gene mutations can result in syndromic diagnosis. With the increasing use of molecular gene analysis in cases of intellectual disability, we may uncover many underdiagnosed cases of Pitt-Hopkins syndrome or other syndromes. Therefore, the scope of the proposed etiology and pathogenesis of intellectual dysfunction can be increased by reporting and tracking more cases.
Availability of data and materials
Not applicable
Abbreviations
- ACMG:
-
American College of Medical Genetics and Genomics
- CASPR2:
-
Contactin-associated protein 2
- CDFE:
-
Cortical dysplasia focal epilepsy
- CNTNAP2 :
-
Contactin-associated protein-like 2 gene
- EEG:
-
Electroencephalography
- NRXN1 :
-
Neurexin 1
- PHS:
-
Pitt-Hopkins syndrome
- TCF4 :
-
Transcription factor 4
- WES:
-
Whole-exome sequencing
References
Pitt D, Hopkins I (1978) A syndrome of mental retardation, wide mouth and intermittent overbreathing. Aust Paediatr J 14:182–184
Peippo M, Simola KO, Valanne LK, Larsen AT, Kahkonen M, Auranen MP et al (2006) Pitt-Hopkins syndrome in two patients and further definition of the phenotype. Clin Dysmorphol 15(2):47–54
de Pontual L, Mathieu Y, Golzio C et al (2009) Mutational, functional, and expression studies of the TCF4 gene in Pitt-Hopkins syndrome. Hum Mutat. 30:669–676
Taddeucci G, Bonuccelli A, Mantellassi I, Orsini A, Tarantino E (2010) Pitt-Hopkins syndrome: report of a case with a TCF4 gene mutation. Ital J Pediatr. 36:12
Zweier C, Sticht H, Bijlsma EK et al (2008) Further delineation of Pitt-Hopkins syndrome: phenotypic and genotypic description of 16 novel patients. J Med Genet. 45:738–744
Whalen S, Heron D, Gaillon T et al (2012) Novel comprehensive diagnostic strategy in Pitt-Hopkins syndrome: clinical score and further delineation of the TCF4 mutational spectrum. Hum Mutat. 33:64–72
Giurgea I, Missirian C, Cacciagli P, Whalen S, Fredriksen T et al (2008) TCF4 deletions in Pitt-Hopkins Syndrome. Hum Mutat 29:E242–E251
Nakabayashi K, Scherer SW (2001) The human contactin-associated protein-like 2 gene (CNTNAP2) spans over 2 Mb of DNA at chromosome 7q35. Genomics 73:108–112
Bellen HJ, Lu Y, Beckstead R, Bhat MA (1998) Neurexin IV, caspr and paranodin—novel members of the neurexin family: encounters of axons and glia. Trends Neurosci 21:444–449
Poot M (2017) Intragenic CNTNAP2 deletions: a bridge too far? Mol Syndromol 8:118–130
Zweier C, de Jong EK, Zweier M, Orrico A, Ousager LB et al (2009) CNTNAP2 and NRXN1 are mutated in autosomal-recessive Pitt-Hopkins like mental retardation and determine the level of a common synaptic protein in Drosophila. Am J Hum Genet 85:655–666
Strauss KA, Puffenberger EG, Huentelman MJ, Gottlieb S, Dobrin SE et al (2006) Recessive symptomatic focal epilepsy and mutant contactinassociated protein-like 2. N Engl J Med 354:1370–1377
Watson CM, Crinnion LA, Tzika A, Mills A, Coates A et al (2014) Diagnostic whole genome sequencing and split-read mapping for nucleotide resolution breakpoint identification in CNTNAP2 deficiency syndrome. Am J Med Genet A 164A:2649–2655
Rodenas-Cuadrado P, Pietrafusa N, Francavilla T, La Neve A, Striano P, Vernes SC (2016) Characterisation of CASPR2 deficiency disorder – a syndrome involving autism, epilepsy and language impairment. BMC Med Genet 17:8
Lee IS, Carvalho CM, Douvaras P, Ho SM, Hartley BJ et al (2015) Characterization of molecular and cellular phenotypes associated with a heterozygous CNTNAP2 deletion using patient-derived hiPSC neural cells. NPJ Schizophr 1:15019
Zollino et al (2019) Diagnosis and management in Pitt-Hopkins syndrome: First international consensus statement. Clin Genet 95:462–478
Depienne C et al (2017) Genetic and phenotypic dissection of 1q43q44 microdeletion syndrome and neurodevelopmental phenotypes associated with mutations in ZBTB18 and HNRNPU. Hum Genet. 136(4):463–479
De W et al (2016) Phenotype and natural history in 101 individuals with Pitt-Hopkins syndrome through an internet questionnaire system. Orphanet J Rare Dis 11:37
Acknowledgements
We gratefully thank the patient’s parents for their cooperation in providing needed information and acceptance to publish the case.
Funding
No funds were received.
Author information
Authors and Affiliations
Contributions
SAl had formulated the diagnostic suspicion of syndromic diagnosis and prepared the case report for publication after being referred from HSh. HSh had defined the clinical picture of the patient (severe psychomotor delay, facial dysmorphisms, and seizures) and formulated the diagnostic suspicion of syndromic diagnosis. The authors have read and approved the manuscript and ensure that this is the case.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The ethical committee in the Welfare Children Hospital had approved the case report to be conducted and published after having a written consent from the child’s mother as the official guardian of the baby.
Consent for publication
A written consent was obtained from the patient’s mother as the official guardian of the baby to publish her child’s case and photo.
Competing interests
Dr. AlBaazi and Dr. Shareef have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
AlBaazi, S., Shareef, H. Case report: Pitt-Hopkins like syndrome with CNTNAP2 mutation. Egypt J Med Hum Genet 21, 70 (2020). https://doi.org/10.1186/s43042-020-00113-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-020-00113-0