
Abstract
Background
Hemophagocytic lymphohistiocytosis (HLH) is a syndrome of pathological immune activation characterized by clinical signs and symptoms of extreme inflammation. It results from the uninhibited proliferation and activation of cells of the macrophage lineage and leads to the production of excess amounts of pro-inflammatory cytokines. The familial form of HLH disease is due to mutations in several genes necessary for natural killer (NK) cell and T cell granule-mediated cytotoxic function. These genes are involved in sorting, trafficking, docking, and fusion of cytotoxic granules containing granzymes A and B and perforin to the cell membrane of the target cell (using the proteins LYST, AP-3 complex, Rab27a, Munc 13–4, Munc 18–2, syntaxin 11). Defect in any of those proteins results in defective cytotoxicity. Consequently, genes included in these steps play valuable roles in the pathogenesis of familial HLH disease including perforin (PRF1) gene in which defect causes familial HLH type 2 (FHL2).
Case presentation
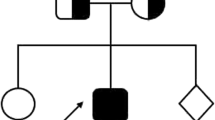
A 2-year-old boy suffered from hepatosplenomegaly and fever. He fulfilled the required criteria for the diagnosis of HLH according to HLH-2004 diagnostic criteria. We screened the patient for the presence of mutations in the coding exons and of PRF1 gene by PCR amplification of genomic DNA followed by direct sequencing of the PCR products. We report a novel homozygous deletion/insertion frameshift mutation in PRF1 gene (M28393: exon 2: c.536delAinsCG p.F178fs). We treated him with HLH 2004 protocol of treatment and showed a remarkable response with resolution of fever and decrement in the size of hepatosplenomegaly.
Conclusions
Our study discovered a novel frameshift mutation in PRF1 gene in an infant with HLH disease, and it is the first report of this type of mutation in Egyptian patients with this disease.
Similar content being viewed by others


Background
Hemophagocytic lymphohistiocytosis (HLH) is a syndrome of pathological immune activation characterized by clinical signs and symptoms of extreme inflammation [1]. Incidence of HLH in Europe and Japan was reported to be 1–2 per million [2], but mostly, it is underestimated due to the difficulties in diagnosis as it has a variable and non-specific symptoms require a high degree of anticipation in both primary and secondary HLH [3]. Histiocyte Society proposed HLH-2004 criteria as a guideline for the diagnosis and treatment of patients with HLH. If the patient has a known genetic defect, the diagnosis of primary HLH (also referred to as familial HLH or FHL) is established. However, for patients without a known positive genetic defect, the diagnosis of either primary or secondary HLH can be done with the presence of at least five out of the eight following diagnostic criteria: fever, splenomegaly, cytopenias [at least bicytopenia; hemoglobin < 9 g/dL (neonates < 10 g/dL), platelet < 100 × 109/L, absolute neutrophil count < 1 × 109/L], hypertriglyceridemia and/or hypofibrinogenemia [fasting serum triglyceride ≥ 3 mmol/L (≥ 265 mg/dL), plasma fibrinogen < 1.5 g/L], serum ferritin > 500 ng/mL, sCD25 (s IL-2 receptor) ≥2400 U/mL, NK cell activity decreased or absent, and hemophagocytosis (bone marrow, other tissues such as lymph nodes, cerebrospinal fluid) [4].
FHL includes five subtypes according to defects in various genes, including chromosome arm 9q mutations (FHL1), PRF1 (FHL2), UNC13D (FHL3), STX11 (FHL4), and STXBP2 (FHL5) [5,6,7,8,9,10,11]. Several mutations are reported in these genes by using molecular genetics approaches to diagnose familial HLH cases.
The overwhelming immune system activation in patients with HLH is related to the genetic defect/s in cases of FHL, while in cases with secondary HLH, it may be due to infection, malignancy, etc. [12].
Case presentation
A 2-year-old boy with a family history of consanguineous marriage and with no documented genetic disease in his family. He was quite well until he showed fever and abdominal enlargement that required medical advice, and therefore, the patient was admitted to the hematology unit in our pediatric hospital for suspicion of malignancy.
On admission, the patient’s temperature was greater than 38.5 °C and hepatosplenomegaly was present (liver span 13 cm, spleen span 11 cm). There were no neurological symptoms. Laboratory studies revealed bicytopenia (hemoglobin 7 g/dL and platelet 13 × 109/L) and notable abnormal findings related to liver function tests. He had an increased level of AST (109 U/L; reference, up to 40 U/L), ALT (116 U/L; reference, up to 41 U/L), LDH (898 U/L; reference interval, 120–300 U/L), total and direct bilirubin (1.1 and 0.8 mg/dL; reference, up to 1 and 0.2 mg/dL respectively), triglyceride level (520 mg/dL; reference interval, 60–160 mg/dL), serum ferritin (> 5000 ng/mL; reference interval, 70–140 ng/mL), and low level of fibrinogen (64 mg/dL; reference interval, 200–400 mg/dL), total protein (5.6 g/dL; reference interval, 6.5–8.5 g/dL), and albumin (2.9 g/dL; reference interval, 3.5–5.2 g/dL). NK function and perforin flow cytometry were not available in our institute. HLH diagnosis was suspected in our patient as he fulfilled five out of the eight required diagnostic criteria according to the HLH-2004 guidelines, but we screened him for serological markers of Epstein-Barr virus (EBV), cytomegalovirus (CMV), hepatitis A, B, and C and all were negative and bone marrow aspiration showed hyper-cellular bone marrow with absent erythroid series without hemophagocytosis. We did not repeat bone marrow aspiration, as our patient fulfilled five out of the eight HLH diagnostic criteria according to the HLH 2004 guidelines, and its repetition would be time-consuming regarding his poor general condition. We screened the patient for the presence of mutations (the screening was not performed for his parents) in the coding exons of PRF1 gene by PCR amplification of genomic DNA, followed by direct sequencing of the PCR products in the following steps:
- 1.
We extracted genomic DNA from fresh peripheral blood using Thermo Scientific Gene JET Whole Blood Genomic DNA Purification Mini kit (Thermo Fisher Scientific Inc., Carlsbad, CA, USA).
- 2.
We used PCR to amplify the coding exons 2 and 3 of the PRF1 gene, including the exon-intron boundaries, using the following primers for exon 2: 5′ CCCTTCCATGTGCCCTGATAATC-3′ and 5′ AAGCAGCCTCCAAGTTTGATTG-39; and exon 3: 5′-CCAGTCC TAGTTCTGCCCACTTAC-3′ and 5′-GAACCCCTTCAGTCCAAG CATAC-3′.
- 3.
We performed amplification of 500 ng of DNA in a 50-μL assay of 25 μL Dream Taq Green PCR Master Mix) (Thermo Fisher Scientific Inc., Carlsbad, CA, USA) 0.4 mmol/L of each primer, and to the rest of volume water.
- 4.
Reaction conditions were 3 min at 95 °C followed by 30 cycles of 45 s at 95 °C, 30 s at 60 °C, 1 min 45 s at 72 °C, and then 10 min at 72 °C.
- 5.
Detection of the amplification product by Agarose gel Electrophoresis.
- 6.
Primers used for sequencing were the same as those for amplification.
- 7.
We performed cycle sequencing using the Big Dye terminator cycle sequencing reaction kit (version 2; Applied Biosystems, Foster City, CA, USA) and separated the DNA fragments on an ABI Prism 3700 DNA Analyzer (Applied Biosystems).
- 8.
We compared sequences with the published PRF1 gene sequence (GenBank accession no. M28393) using MEGA software (Molecular Evolutionary Genetic Analysis) which is a powerful sequence analysis software package, Basic Local Alignment Search Tool (BLAST) on The National Center for Biotechnology Information (NCBI), and Chromas software.
Results revealed a novel homozygous frameshift mutation in PRF1 gene (M28393: exon 2: c.536delAinsCG p.F178fs) with the production of stop codon as shown in Figs. 1, 2, and 3.

Gene mutation causes a frameshift leading to an early termination/stop codon using Chromas software

Gene mutation causes a frameshift leading to an early termination/stop codon using BLAST at NCBI

Protein translation shows resultant stop codon using MEGA software
We treated our patient medically with the HLH-2004 protocol including etoposide, dexamethasone, and cyclosporine A. He showed dramatic responses with a resolution of fever, decrement in size of hepatosplenomegaly, and correction of hepatitis and cytopenia. We plan for hematopoietic stem cell transplantation (HSCT), and he is on the waiting list as HSCT is not available in our institute.
Discussion
The clinical findings of FHL are usually non-specific, and in the majority of these patients, fever and splenomegaly are present from the onset of the disease. Without treatment, nearly all HLH patients develop severe pancytopenia [13]. The presence of cytopenia can be explained by two factors: the first is hemopoiesis suppression by the highly elevated levels of inflammatory cytokines, and the second is the phagocytosis of blood cells by over-activated macrophages [14]. Hemophagocytosis in the bone marrow may not be present early in the disease and could be detected in half to two-thirds of HLH patients by repetition of BM examination [15, 16].
In our patient, the PRF1 gene mutation c.536delAinsCG p.F178fs was found which is an interesting finding as to the best of our knowledge, this is the first reported case in Egypt with deletion and insertion mutation as known PRF1 gene mutations in Egypt are either missense or non-sense mutation [17].
About 20% of familial cases of HLH are caused by PRF1 gene mutations, with high incidence in North America (approximately 50%), Japan (40%), and Turkey (30%) [7, 13].
Several studies identified that certain PRF1 gene mutations are associated with lymphomas, type 1 diabetes mellitus, acquired aplastic anemia, autoimmune lymphoproliferative syndrome (ALPS), and multiple sclerosis [18].
For PRF1 gene, more than 120 different mutations have been detected: around 100 missense/non-sense mutations and 21 deletion/insertion mutations [17]. Some PRF1 gene mutations are more common in particular ethnic populations, suggesting a common ancestry. For example, the Trp374 stop (G1122A) mutation occurs frequently in Turkish families, while the Leu17 frameshift (50delT) mutation occurs frequently in African populations [19].
Familial HLH type 2 (FHL 2) which is considered a lethal childhood disease is caused by losing perforin activity [8], so molecular diagnosis and initiation of management for FHL2 is life-saving [15]. Immunochemotherapy-based treatments can result in remission, but a relapse may occur, making HSCT the only curative treatment for FHL [20].
Conclusions
A novel mutation in PRF1 gene was detected in our patient with FHL2 disorder. Our study may help to apply appropriate genetic counseling and prenatal diagnosis for individuals at higher risk of HLH disorder.
Since there are high rate first-degree consanguineous marriages in our country, finding and reporting rare novel mutations would be very important for the prevention of FHL2 with a homozygous pattern of inheritance. Therefore, PRF1 gene molecular genetic testing for families of patients diagnosed as FHL2 is important.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ALPS:
-
Autoimmune lymphoproliferative syndrome
- BLAST:
-
Basic Local Alignment Search Tool
- CMV:
-
Cytomegalovirus
- DNA:
-
Deoxyribonucleic acid
- EBV:
-
Epstein-Barr virus
- FHL:
-
Familial hemophagocytic lymphohistiocytosis
- HLH:
-
Hemophagocytic lymphohistiocytosis
- HSCT:
-
Hematopoietic stem cell transplantation
- NCBI:
-
National Center for Biotechnology Information
- NK:
-
Natural killer
- PCR:
-
Polymerase chain reaction
- PRF1 :
-
Perforin
- sHLH:
-
Secondary HLH
References
Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain KL (2011) How I treat hemophagocytic lymphohistiocytosis. Blood 118(15):4041–4052
Contino A, Trombatore G, Timeus F. Hemophagocytic lymphohistiocytosis in pediatric patients: a review. of. 2018;5:35-40.
Ramachandran S, Zaidi F, Aggarwal A, Gera R (2017) Recent advances in diagnostic and therapeutic guidelines for primary and secondary hemophagocytic lymphohistiocytosis. Blood Cells Mol Dis 64:53–57
Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S et al (2007) HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 48(2):124–131
Côte M, Ménager MM, Burgess A, Mahlaoui N, Picard C, Schaffner C et al (2009) Munc18-2 deficiency causes familial hemophagocytic lymphohistiocytosis type 5 and impairs cytotoxic granule exocytosis in patient NK cells. J Clin Invest. 119(12):3765–3773
Gholam C, Grigoriadou S, Gilmour K, Gaspar H (2011) Familial haemophagocytic lymphohistiocytosis: advances in the genetic basis, diagnosis and management. Clin Experiment Immunol 163(3):271–283
Stepp SE, Dufourcq-Lagelouse R, Le Deist F, Bhawan S, Certain S, Mathew PA et al (1999) Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. 286(5446):1957–1959
Voskoboinik I, Smyth MJ, Trapani JA (2006) Perforin-mediated target-cell death and immune homeostasis. Nat Rev Immunol 6(12):940
Zur Stadt U, Rohr J, Seifert W, Koch F, Grieve S, Pagel J et al (2009) Familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) is caused by mutations in Munc18-2 and impaired binding to syntaxin 11. Am J Human Genet 85(4):482–492
Zur Stadt U, Schmidt S, Kasper B, Beutel K, Diler AS, Henter J-I et al (2005) Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type-4 to chromosome 6q24 and identification of mutations in syntaxin 11. Human Mol Genet 14(6):827–834
Feldmann J, Callebaut I, Raposo G, Certain S, Bacq D, Dumont C et al (2003) Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell. 115(4):461–473
Hines M, Bhatt N, Talano JAM. Diagnosis, treatment, and management of hemophagocytic lymphohistiocytosis in the critical care unit. Critical Care of the Pediatric Immunocompromised Hematology/Oncology Patient: Springer; 2019. p. 159-82.
Morimoto A, Nakazawa Y, Ishii E (2016) Hemophagocytic lymphohistiocytosis: pathogenesis, diagnosis, and management. Pediatr Int. 58(9):817–825
Zhang Z, Wang J, Ji B, Greenwood B, Zhang Y, Wang Y et al (2016 Apr) Clinical presentation of hemophagocytic lymphohistiocytosis in adults is less typical than in children. Clinics. 71(4):205–209
Ishii E, Ohga S, Imashuku S, Yamamoto K, Yasukawa M (2005) Review of familial hemophagocytic lymphohistiocytosis (FHL) in children with focus on Japanese experiences. Crit Rev Oncol Hematol. 53:209–223
Risma KA, Marsh RA (2019) Hemophagocytic lymphohistiocytosis: clinical presentations and diagnosis. J Allergy Clin Immunol Pract. 7(3):824–832
Sieni E, Cetica V, Hackmann Y, Coniglio ML, Da Ros M, Ciambotti B et al (2014) Familial hemophagocytic lymphohistiocytosis: when rare diseases shed light on immune system functioning. Front Immunol 5:167
Willenbring R, Johnson A (2017) Finding a balance between protection and pathology: the dual role of perforin in human disease. Int J Mol Sci 18(8):1608
Lee SM, Sumegi J, Villanueva J, Tabata Y, Zhang K, Chakraborty R et al (2006) Patients of African ancestry with hemophagocytic lymphohistiocytosis share a common haplotype of PRF1 with a 50delT mutation. J Pediatrics. 149(1):134–137
Jia C, Wang B, Zhu G, Zhang R, Wang K, Yan Y et al (2018) Haploidentical hematopoietic stem cell transplantation using reduced-intensity conditioning for pediatric patients with familial hemophagocytic lymphohistiocytosis. Pediatr Invest 2(4):216–221
Acknowledgements
We would like to thank the patient’s family for their willingness to take part in this study.
Funding
Not applicable
Author information
Authors and Affiliations
Contributions
MA conceived and designed the study, collected, assembled and interpreted the data, and wrote the manuscript. EGB clinically evaluated the patient’s interpreted data and wrote the manuscript. AEF performed the experiments and helped with writing of the manuscript. SHS helped critically in revising the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Approval was obtained from the Zagazig University Institutional Review Board (ZU-IRB #3939).
Consent for publication
Written consent was obtained to participate in this study and to allow us to publish the result of study.
Competing interests
Not applicable
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Almalky, M., Saleh, S.H.A., Baz, E.G. et al. Novel mutation in perforin gene causing familial hemophagocytic lymphohistiocytosis type 2 in an Egyptian infant: case report. Egypt J Med Hum Genet 21, 24 (2020). https://doi.org/10.1186/s43042-020-00067-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-020-00067-3