
Abstract
The study of adrenal disorders is a key component of scientific research, driven by the complex innervation, unique structure, and essential functions of the adrenal glands. This review explores the use of non-traditional animal models for studying congenital adrenal hyperplasia. It highlights the advantages, limitations, and relevance of these models, including domestic ferrets, dogs, guinea pigs, golden hamsters, pigs, and spiny mice. We provide a detailed analysis of the histological structure, steroidogenesis pathways, and genetic characteristics of these animal models. The morphological and functional similarities between the adrenal glands of spiny mice and humans highlight their potential as an important avenue for future research.
Similar content being viewed by others
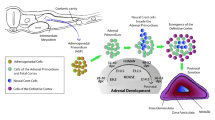

Background
The adrenal glands have always been a subject of scientific interest due to their heterogeneous structure, the variety of hormones that adrenal glands synthesise, complex innervation, and a multitude of physiological functions. They were first mentioned by Bartolomeo Eustachi, who described the adrenal glands as “glandulae quae renibus presentative” (glands located near the kidneys) in his book Opuscola Anatomica, published in 1564 [1]. Up until the mid-19th century, their study was limited to certain anatomical investigations, until Thomas Addison in 1855 discovered that their dysfunction leads to a clinical syndrome, now named after him [1]. However, despite the substantial amount of research and data accumulated on adrenal glands, this organ remains a subject of scientific interest, especially in the context of the search for new methods of diagnosis and treatment of some adrenal diseases.
For example, one form of primary adrenal insufficiency is congenital adrenal hyperplasia (CAH), a group of autosomal recessive disorders with an overall prevalence of 1: 9498 live births in world [2]. Pathogenesis is based on defects in enzymes or transport proteins involved in the biosynthesis of steroid hormones in the adrenal cortex [3]. Insufficiency arises from mutations in genes that encode enzymes of various steps in the biosynthesis of steroid hormones, leading primarily to decreased cortisol synthesis and hyperproduction of adrenocorticotropic hormone (ACTH) due to altered negative feedback loops. As a result, adrenal cortex hyperplasia develops and the target hormone and precursor accumulate above the enzymatic block [3, 4].
Currently, the predominant therapeutic approach to address CAH involves the administration of lifelong replacement hormonal therapy. This therapy serves the purpose of mitigating endogenous hormone deficiency while simultaneously suppressing the secretion of corticotropin-releasing hormone and ACTH, thus restoring the perturbed feedback system through exogenously supplied hormonal agents adrenocorticotropic hormone [4]. However, despite this, this therapeutic modality has no limitations. For example, the administration of hormones based on exogenous tablets poses challenges in the context of infants and very young children. Furthermore, the therapeutic regimen fails to faithfully recapitulate the intricate physiological diurnal rhythm of hormone secretion or response to stressors. The potential stunting of growth in paediatric patients subjected to routine glucocorticoid therapy is of note. The process of determining the optimal dose requires extensive and sustained adjustments, and patient adherence may not always be reflected in the intended therapeutic outcome. Rehabilitation of disrupted negative feedback mechanisms may be suboptimal in certain cases, examples, highlighting a pertinent issue [5]. Furthermore, prolonged administration of glucocorticoids, especially at supraphysiological levels, is associated with the emergence of adverse effects that include obesity, insulin resistance, sarcopenia, dermal atrophy, and other sequelae [6, 7].
In recent years, efforts have been made to develop new glucocorticoid delivery systems or alternative therapeutic approaches that do not involve glucocorticoids. However, the question of fully correcting the symptoms of adrenal insufficiency remains relevant [8]. A radically new approach to treating this condition is based on genetic and cellular technologies. Currently, there is active development in genome editing tools delivery systems, various approaches to gene cellular therapy, gene cellular, and protocols for the culture of functional adrenal cortex cells in vitro [9].
Advancement of genetic and cellular treatment techniques requires preliminary investigations conducted in animal models, often using mice and rats. However, an investigation by Marjut Pihlajoki et al. underscores the substantial morphological and functional disparities between the adrenal glands of these animals and those of humans [10]. There is no zona reticularis (ZR) in the adrenal gland of mice and rats and therefore there is no androgen production [10]. In addition, in rats, there is an undifferentiated zone located in the cortex, which is situated between the zona glomerulosa (ZG) and zona fasciculata (ZF) [11]. It has been suggested that stem and/or progenitor cells expressing steroidogenic factor-1, which has been confirmed by immunohistochemistry study [11]. In contrast to the ZR, young mice exhibit an X-zone that disappears upon reaching sexual maturity in males and during pregnancy or within 3–7 months postpartum in the absence of pregnancy in females [10, 12]. This review will describe alternative animal models that could be used to study CAH.
Main text
The structure of the human adrenal glands and steroidogenesis
To foster a more comprehensive grasp of the distinctive strengths and constraints inherent to each model, a brief elucidation is warranted concerning the configuration of human adrenal glands and the process of steroidogenesis before embarking on the discourse surrounding alternative animal models for the examination of CAH.
The adrenal glands are paired parenchymal organs of the endocrine system, consisting of the adrenal medulla and adrenal cortex, each of which performs specific functions and has different origins [13].
The adrenal medulla develops from neural crest cells and is located within the ZR of the adrenal cortex and is made up of three types of cells: chromaffin cells (pheochromocytes), ganglion cells, and supporting cells [14,15,16]. Chromaffin cells are the predominant cell type in the adrenal medulla, forming clusters of cells that synthesise catecholamines (norepinephrine and epinephrine). Supporting cells are located around these clusters, while ganglion cells are dispersed individually or form clusters between chromaffin cells or near nerve fibres [17, 18].
The adrenal cortex develops from the embryonic mesoderm and synthesises steroid hormones. The adrenal cortex is encased in a fibrous capsule, also derived from the mesoderm, which serves protective and supportive functions and acts as a reservoir for stem cells from the adrenal cortex [19, 20]. In the context of humans, the adrenal cortex manifests three discernible zones characterised by different morphological and functional attributes: ZG, ZF, and ZR [20].
The outermost layer, known as ZG, represents approximately 5–15% of the total cortical volume. It acts as the primary site for the biosynthesis of mineralocorticoids, predominantly aldosterone. This vital hormone plays a crucial role in the meticulous regulation of potassium and sodium levels in the bloodstream [21].
Below the ZG lies the ZF, which occupies up to 75% of the total volume of the cortex and synthesises glucocorticoids, primarily cortisol, together with a smaller number of androgens [22]. Glucocorticoids play a crucial role in the body’s stress response, influencing metabolism, particularly carbohydrate metabolism, and to a lesser extent electrolyte balance due to some mineralocorticoid activity [23,24,25,26].
The innermost layer of the cortex, recognised as ZR, represents approximately 10–20% of the total cortical volume. Within this region, the synthesis of steroid hormones occurs, encompassing mildly active androgens such as DHEA, DHEA-S, androstenedione, androstenediol, 11β-hydroxyandrostenedione, more biologically active androgens like 11β-hydroxytestosterone and testosterone, in addition to a small amount of glucocorticoids [13, 22, 27].
The process of steroid hormone biosynthesis in the adrenal cortex begins with cholesterol and is subject to regulation by the adrenocorticotropic hormone (ACTH) secreted by the anterior pituitary (Fig. 1). Cholesterol can be obtained from dietary sources, particularly those of animal origin. Furthermore, within the adrenal glands and ovaries, cholesterol can be synthesised de novo from acetate. Furthermore, a proportion of cholesterol is manufactured within the liver, predominantly in the form of low-density lipoproteins [28, 29].

Scheme of steroidogenesis and CAH types. StAR -steroidogenic acute regulatory protein; 3β-HSD − 3 β -hydroxysteroid dehydrogenase; 17β -HSD − 17β -hydroxysteroid dehydrogenase; DHEA - dehydroepiandrosterone; SULT 2A1 - sulfotransferase
The beginning of steroid hormone synthesis is induced by the acute steroidogenic regulatory protein (StAR), which facilitates the translocation of cholesterol from the cytoplasm to the outer and, subsequently, inner mitochondrial membranes [30, 31]. Within mitochondria, cholesterol undergoes the conversion to pregnenolone under the catalytic influence of the enzyme 20,22-desmolase (encoded by the CYP11A1 gene). This transformation involves a series of successive hydroxylation reactions, culminating in the cleavage of the cholesterol side chain. Once pregnenolone is generated, which serves as the precursor of all steroid hormones that include mineralocorticoids, glucocorticoids, androgens, estrogens, and progesterone, it is translocated from the mitochondria to the endoplasmic reticulum [32].
Pregnenolone undergoes a 17α-hydroxylation reaction catalysed by the 17-hydroxylase enzyme 17α-hydroxylase to form 17-hydroxypregnenolone, which can then be converted to progesterone by the enzyme 3β-hydroxysteroid dehydrogenase (3β-HSD). The 17α-hydroxylase catalyses carbon atom hydroxylation at position 17, while 17,20-lyase catalyses side chain cleavage at positions 17 and 20, leading to the formation of 19-carbon precursors of sex steroids. The enzyme 3β-HSD converts pregnenolone to progesterone and DHEA to androstenedione, and the enzyme 17β-hydroxysteroid dehydrogenase (17β-HSD) is involved in the formation of testosterone from androstenedione [33]. In the absence of 17-hydroxylase activity (for example, in the ZG of the adrenal cortex), pregnenolone is converted to mineralocorticoids. If 17α-hydroxylase activity is present in the absence of 17,20-lyase activity (for example, in the ZF of the adrenal cortex), pregnenolone is converted to cortisol through the glucocorticoid pathway [33].
Classification and etiology of congenital adrenal hyperplasia (CAH)
Before delving into the discussion of non-classical animal models, it is worth mentioning a few words about the classification and clinical manifestations of CAH. Currently, seven clinical genetic variants of CAH are identified (Fig. 1) [34]:
-
1.
Lipoid hyperplasia of the adrenal cortex (StAR gene defect).
-
2.
Lipoid hyperplasia of the adrenal cortex − 20,22-desmolase deficiency (CYP11A1 gene defect).
-
3.
17α-hydroxylase/17,20-lyase deficiency (CYP17A1 gene defect).
-
4.
3β-hydroxysteroid dehydrogenase deficiency (HSD3B2 gene defect).
-
5.
21-hydroxylase deficiency (CYP21A2 gene defect).
-
6.
11β-hydroxylase deficiency (CYP11B1 gene defect).
-
7.
Oxidoreductase deficiency (POR gene defect).
Lipoid hyperplasia of the adrenal cortex develops due to a defect in the StAR gene located on the short arm of chromosome 8 (8p11.2) or a defect in the CYP11A1 gene located on chromosome 15 (15q23-24) [35,36,37]. Deficiency of the StAR protein, as well as 20,22-desmolase, leads to the development of a severe form of CAH characterised by the absence of synthesis of all classes of steroids in both the adrenal glands and the gonads (ovaries, testes) [38, 39].
CAH caused by defects in the CYP17A1 gene, located on chromosome 10 (10q24.3), is characterised by a deficiency of both 17α-hydroxylase and 17,20-lyase [40]. The lack of these enzymes leads to a deficiency of glucocorticoids and androgens. Deficiency of 17α-hydroxylase disrupts cortisol synthesis, leading to ACTH hyperproduction and activation of aldosterone precursor synthesis, while deficiency of 17,20-lyase leads to impaired androgen synthesis in adrenal glands and gonads [41, 42]. Mutations in the CYP17A1 gene are relatively rare, with several mutations reported that cause complete or combined deficiency of the 17-hydroxylase / 17,20-lyase enzyme or isolated 17,20-lyase deficiency [43,44,45,46,47].
The deficiency of 3β-hydroxysteroid dehydrogenase type 2 3-hydroxysteroid dehydrogenase (HSD3B2) is extremely rare and is caused by mutations in the HSD3B2 gene located on chromosome 1 (1p12) [47, 48]. It is characterised by impaired synthesis of all classes of steroid hormones in the gonads and adrenal glands [48,49,50,51].
The most common form of CAH, accounting for more than 95% of cases, is due to a deficiency of the enzyme 21-hydroxylase, resulting from mutations in the CYP21A2 gene located on chromosome 6 (6p21.33) [52, 53]. The CYP21A2 gene is located in tandem with a highly homologous pseudogene, CYP21A1P. Unequal crossover between these genes during meiosis leads to deletions and the formation of non-functional chimeric genes [54]. The 21-hydroxylase enzyme belongs to the cytochrome P450 monooxygenase family, is located in the cisterns of the endoplasmic reticulum and is involved in the conversion of progesterone and 17-hydroxyprogesterone to 11-deoxycorti-costerone and 11-deoxycortisol, respectively [55,56,57,58]. Depending on the amount of the active 21-hydroxylase enzyme, two clinical forms of 21-hydroxylase deficiency are distinguished: classical and non-classical. The classical form, observed in approximately 75% of cases, is characterised by low enzyme activity, resulting in a deficiency of not only cortisol, but also aldosterone and excess testosterone production due to the diversion of precursor hormones toward androgen production. The non-classical form develops with preserved enzyme activity at 20–50%, characterised by a less significant reduction in cortisol production and excessive androgen synthesis after puberty [59, 60].
Deficiency of 11β-hydroxylase arises from a mutation in the CYP11B1 gene located on chromosome 8q21 [61]. This enzyme catalyses the addition of hydroxyl groups, converting 11-deoxycortisol and 11-deoxycorticosterone into cortisol and corticosterone, respectively [62]. Deficiency of 11β-hydroxylase disrupts cortisol synthesis, which, through a negative feedback mechanism, stimulates ACTH synthesis and leads to the formation of cortisol and aldosterone precursors, as well as androgens [63]. Furthermore, 11-deoxycorticosterone has mineralocorticoid activity and its accumulation leads to the development of arterial hypertension, sometimes with a malignant course that cannot be alleviated with medication [64].
P450 oxidoreductase (POR) is a relatively rare form of CAH, first described in 2004 [65]. This form of CAH is caused by mutations in the POR gene, located on chromosome 7 (7q11.2), responsible for the formation of the enzyme P450 oxidoreductase. This enzyme is necessary to provide oxygen molecules to all microsomal enzymes in the cytochrome P450 family, including the previously mentioned CYP17A1 and CYP21A2 [66]. Patients with POR deficiency exhibit a combined partial deficiency of 17α-hydroxylase and 21-hydroxylase deficiency, which clinically often manifests itself as moderately reduced levels of glucocorticoid and mineralocorticoid, as well as elevated androgen concentrations in the blood [67].
According to the literature, CAH is rare in animals. One case of CAH has been described in a cat [66]. A mutation in a gene similar to 11β-hydroxylase is associated with clinical manifestations such as polyuria, polydipsia, and increased blood pressure [68].
Non-classical emerging animal models
Nonclassical animal models that have the potential to be studied include ferrets, dogs, guinea pigs, golden hamsters, pigs, primates, and spiny mice (Fig. 2).

Comparative characteristics of non-classical animal models. ZG – zona glomerulosa; IZ – intermediate zone; ZF – zona fasciculata; ZR – zona reticularis; JZ - juxtamedullary zone; M – medulla
Domestic ferret or ferret
According to the work of Holmes, RL, who first described the structure of adrenal glands in domestic ferrets, their adrenal glands are encapsulated in a thick connective tissue capsule, under which two well-distinguishable parts are identified: the cortex and medulla. Three main zones characteristic of the human adrenal cortex, ZG, ZF, and ZR, are easily distinguishable from each other [69]. Additionally, there are other structures that are not observed in the human adrenal glands. For example, between the ZG and the ZF, an irregularly shaped narrow strip is identified that forms an intermediate zone. At the cellular level, this structure is made up of small cells of irregular shaped with centrally located nuclei. Furthermore, between the ZR and the medulla, a juxtamedullary zone is present [69]. Similarly to human physiology, mineralocorticoids are synthesised in the ZG, glucocorticoids, including cortisol in the ZF, and androgens such as estradiol, 17-hydroxyprogesterone, and androstenedione in the ZR [70].
Within the medulla, prominent chromaffin cells of considerable size can be discerned, characterised by a centrally positioned spherical nucleus and a pale cytoplasm replete with granular elements. Furthermore, clusters of ganglion cells manifest their presence, occupying positions within the confines of the medulla or at the junction that delimits the cortex and medulla [71].
An advantage of domestic ferrets as a model for studying adrenal diseases is their expression of the CYP17A1 gene, which is responsible for the synthesis of enzymes that catalyse both the 17α-hydroxylation reaction necessary for cortisol production and the 17,20-lyase reaction necessary for androgen synthesis [71, 72]. Cells in ZF and ZR possess 17α-hydroxylase activity, suggesting that cortisol is the main glucocorticoid in this species, similar to humans [73, 74]. It should be noted that androgen synthesis in the adrenal glands of domestic ferrets is limited [75]. This is due to restricted synthesis of the cytochrome B5 enzyme (Cyt-b5), an allosteric enzyme that regulates the activity of 17,20-lyase, which is necessary for androgen synthesis in the adrenal glands [72].
In addition, domestic ferrets serve as a valuable experimental model for a comprehensive investigation of the intricate mechanisms that underlie the process of tumorigenesis within the adrenal cortex. Compared to specific murine species, ferrets subjected to gonadectomy exhibit the emergence and progression of neoplastic growths within the confines of the adrenal cortex, as documented in the study by reference [73]. It is of particular significance to emphasise that cellular entities that originate within the boundaries of the adrenal cortex during the course of this pathological progression demonstrate a predilection for the secretion of androgenic hormones, in contrast to the synthesis and release of mineralo- and glucocorticoids [75]. This biosynthetic activity centred on androgen production within cortical tissue precipitates a physiological state characterised and identified as adrenal-associated endocrinopathy. The clinical manifestations that follow encompass bilateral symmetric alopecia and hypertrophy of the external genitalia, as elucidated in scholarly discourse [76]. Confirmation and validation of this diagnostic categorisation is established through the discernible elevation of circulating concentrations of 17α-hydroxyprogesterone, androstenedione, DHEA-S, or estradiol within the plasmatic environment, as expounded in the publications denoted by references [75, 76].
However, despite the structural parallels that exist between the adrenal glands of domestic ferrets and humans, coupled with the shared expression of the CYP17A1 gene and the subsequent androgen biosynthetic processes, certain limitations persist that curtail the extensive applicability of these creatures as a suitable experimental paradigm. Among the shortcomings inherent in this model are the challenges associated with the purchase of an adequate cohort of animals, the inherent genetic heterogeneity prevalent within the population, distinctive behavioural attributes, and the imposition of more intricate requirements related to laboratory housing conditions, in stark contrast to comparatively well-established and available model organisms such as rats and mice, as extensively elaborated in the scholarly discourse presented by references [77, 78].
Dogs
Dogs can also be considered emerging models for studying adrenal diseases. In the adrenal cortex of dogs, similar to that of humans, three zones are distinguished: ZG, ZF, and ZR [79]. Like other mammals, the ZG regulates sodium and potassium levels in blood plasma, while the ZF and ZR together function as the main source of glucocorticoid secretion [13]. Furthermore, androgens such as DHEA and androstenedione are produced in the ZR [80].
Given the commonality of the end products of adrenal cortical synthesis, specifically aldosterone and cortisol, shared between humans and dogs, the assumption was established that the steroidogenesis processes would exhibit parallelism in both species. In recent literature, the sequence of adrenal cortical steroidogenesis in dogs closely parallels the established human steroidogenesis pathway [81].
In humans, the final stages of aldosterone and cortisol synthesis, as well as the final stages of aldosterone and corticosterone synthesis in rats and mice, are catalysed by two different related enzymes: aldosterone synthase and 11β-hydroxylase, which are encoded by the genes CYP11B2 and CYP11B1, respectively [82, 83]. However, according to the NCBI database, the dog genome contains CYP11B2 (NC_006595.3) but not CYP11B1.
Sanders et al. show that there is a significant gap in the canine genome near the known CYP11B2 gene sequence and that the CYP11B1 gene is absent in dogs, which should have been involved in steroidogenesis [84]. Sanders et al. also suggested an alternative scheme of steroidogenesis in dogs without the use of aldosterone synthase [85]. Similar to ferrets, androgen synthesis under physiological conditions is limited in dogs [75, 85]. The reasons for this phenomenon are unknown [86].
Despite the identical structure of the adrenal cortex in dogs and humans, as well as the same end products of synthesis, there are differences in the catalysts of the reaction, one notable example being the absence of aldosterone synthase [84]. Additionally, due to their size and storage conditions, dogs are less commonly used in laboratory research compared to smaller animals with simpler housing conditions, making them a questionable model for studying human adrenal diseases.
Guinea pigs
Guinea pigs, classified as domesticated rodents that fall under the genus Cavia and the Caviidae family, are a laudable model for laboratory animal studies due to their body mass, dimensions, ease of manipulation and care prerequisites [87, 88].
The adrenal glands of guinea pigs are covered by a connective tissue capsule, under which the cortex and medulla are distinguished. According to the literature, the adrenal cortex histologically reveals three zones: the ZG, the ZF and the ZR [89, 90]. However, according to Sheikhian A. and others, an intermediate zone is identified between the ZG and the ZF, composed of a thin layer of small irregularly shaped cells with dark stained nuclei. Furthermore, the cortex extends deep into the medulla, revealing structures reminiscent of invaginations [91]. In the study by Lafi A. and colleagues, the ZF of the adrenal cortex is composed of two morphologically distinguishable segments: the outer section comprises polygonal cells organised in parallel bundles, while the inner segment is composed of cells arranged in irregular cords, interspersed with sinuses [89]. Histologically, the medulla contains chromaffin and ganglion cells [89,90,91].
Immunohistochemical staining of guinea pig adrenal glands using antibodies against three enzymes revealed that the expression of 3β-HSD, 20,22-desmolase is present in all zones of the adrenal cortex, while the expression of 17α-hydroxylase is observed only in the ZF and ZR. The staining was more intense in the outer layer of the ZF and gradually decreased toward the inner layers of the ZF and ZR [92]. The results indicate that the 17-hydroxylase enzyme 17α-hydroxylase is synthesised in these animals, resulting in the formation of cortisol and androgens [92, 93]. It should be noted that steroidogenesis in the adrenal glands of guinea pigs occurs differently than in humans [94]. For example, androgens DHEA and DHEA-S are not detected in guinea pig blood, but another C19 steroid, 11β-hydroxyandrostenedione, is present [95,96,97].
Despite their near-ideal status as laboratory animals, the configuration of their adrenal glands and variations in the synthesis of specific steroid hormones hinder the use of guinea pigs as an optimal model for investigating adrenal diseases.
Cricetidae
The Syrian hamster, known colloquially as the golden hamster, belongs to the rodent family Cricetidae and is one of the most popular species among domesticated animals [98].
Researchers have been interested in the adrenal glands of these animals since the mid-20th century, studying not only their histological structure, but also determining the spectrum of hormones they produce [99,100,101,102,103].
Similarly, for most mammals, the adrenal glands of the Syrian hamster are enveloped by a thin connective tissue capsule, beneath which the cortex and medulla are discernible [102]. The adrenal cortex can be divided into three zones: ZG, ZF, and ZR [91, 101]. In particular, these animals synthesise the enzyme 17α-hydroxylase, 3β-HSD, which participates in the formation of androgens and cortisol [104,105,106]. The medulla is made up of chromaffin cells [107].
The benefits of this model include its similarity to human adrenal glands in terms of both morphology and function, as well as its relatively simple laboratory maintenance requirements.
Recent study has uncovered significant heterogeneity among several members of the Cricetidae family, directly linked to their genetic characteristics. In particular, a new cortical region, zona inaudita, was identified in the Oldfield mouse (Peromyscus polionotus), which expresses the enzyme AKR1C18. This enzyme converts progesterone into 20α-hydroxyprogesterone [108]. We expect this discovery to continue attracting the attention of researchers in the field in the near future, as it opens up new possibilities for understanding the complex genetic underpinnings of these animals.
Pigs
Large animals such as pigs are considered nearly ideal animals for modelling various adrenal gland diseases due to their anatomical and physiological similarity to the human paired organ [109,110,111]. These model animals offer opportunities for the detection of biomarkers during pathological conditions and the study of new drugs [112, 113].
Similarly to humans, the adrenal cortex of pigs consists of three morphological layers: the ZG, the ZF, and the ZR [114]. The end products of steroidogenesis are identical to those of humans.
Twinkle Vohra’s research suggests that pigs could be used as a suitable model for studying primary hyperaldosteronism, due to their similarities with humans [115]. However, rodents lack certain genetic expressions, such as KCNJ5, which limits their ability to mimic this condition [115]. On the other hand, pigs exhibit comparable potassium channel gene profiles, overcoming this obstacle.
A limitation of using pigs as animal models to study human adrenal gland diseases is the expression of only one enzyme, CYP11B1, in the adrenal cortex, while humans also have CYP11B2. Additionally, their larger size and complex housing requirements can be considered disadvantages [116].
Primates
Primates are important laboratory animals for biomedical, pharmacological and toxicological research. Several taxonomic groups of primates are used in research, including prosimians (e.g. lorises and lemurs), New World monkeys (e.g. marmosets and capuchins), Old World monkeys (e.g. macaques and baboons) and apes (e.g. gibbons, gorillas, orangutans and chimpanzees) and some of them have their own special features [117]. The adrenal glands are very similar to the human adrenal glands: they also have 3 different zones in the cortex and produce androgens, but there are exceptions [117].
A study by Toru Tachibana et al. [118] compared two species of New World monkeys: Aotus spp. (night monkeys) and Saimiri spp. (squirrel monkeys). Both species have a similar adrenal structure, but the adrenal cortex of Aotus spp. showed a significant enlargement of the ZF, and the fasciculata cells showed a remarkable accumulation of large lipid droplets and irregular shapes of mitochondrial cristae. However, serum cortisol and ACTH levels were lower in Aotus than in Saimiri [118].
Three zones of the adrenal cortex have been identified in Rhesus macaques. Expression of SULT2A1 and cytochrome b5 has been detected in the ZR. This indicates active androgen production in this region. Similar to humans, expression of 3β-HSD was not observed in this study [119, 120]. The enzyme 17α-hydroxylase is also present in the Rhesus macaques [121].
However, Patterson et al. found that newborn male marmosets have a fetal zone that produces androgens, similar to humans. In contrast, adult male marmosets do not appear to have an actively functioning androgen-producing ZR [121].
Despite the fact that the adrenal glands of primates and humans are highly similar, this model has limitations due to the conditions of confinement, the complexity of interacting with these animals, and the costs associated with conducting research.
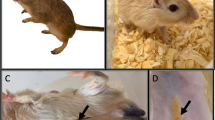
Spiny mice
Mice of the genus Acomys, commonly known as spiny mice, are rodents that inhabit the deserts of the Middle East, Africa, and South Asia, known for their complex social organisation. They are named for the prominent spiky hairs on their backs [122]. Spiny mice belong to the Muridae family, which comprises almost a third of all rodent diversity [123]. Unlike other rodents, where organ development completes after birth, many internal organs in spiny mice develop during the embryonic period [124]. This has led some researchers to propose the use of spiny mice as models for embryogenesis [125]. Young spiny mice are highly mobile compared to neonates from other laboratory rodents, and they can consume solid food from the second day of life [126].
Spiny mice have been used as research animals since at least 1911, but interest in them intensified in 2012 when their regenerative capabilities were first discovered [126, 127]. Spiny mice were used as models for the study of diabetes, as they showed spontaneous Langerhans islet degeneration, a tendency toward hyperglycemia, and obesity-related diabetes, but without insulin resistance [128]. Furthermore, physiological studies were performed to assess kidney function, which revealed a high concentration of urea in the urine, possibly due to habitat [129]. Bellofiore et al. described spiny mice as the first rodents to exhibit a menstrual cycle that lasts 8–9 days [130].
The study of spiny mouse adrenal glands is relatively recent, but it is already known that they are more similar to human adrenal glands compared to the more usual laboratory mouse and rat models [10]. The adrenal glands of spiny mice are covered by a connective tissue capsule and contain the cortex and medulla. In the cortex, three sequential zones are distinguished: ZG, ZF, and ZR. Furthermore, immunohistochemical staining has shown the expression of 17α-hydroxylase, 3β-HSD, CYB5a in the cortex on day 30 of gestation, which is necessary for cortisol and androgen synthesis [131, 132]. In a study by Lamers W. H. et al., cortisol was found to be the primary glucocorticoid in spiny mice, while corticosterone is dominant in rats [125]. Another study (Quinn TA et al.) identified chromaffin cells in the medulla using antibodies to tyrosine hydroxylase, as well as presumptive sympathetic neurones detected by antibodies to synaptophysin and nonphosphorylated neurofilament H [131].
The advantages of this model include its similar structure to the human adrenal glands, both morphologically and functionally, as well as its simple laboratory maintenance requirements.
Conclusions
The animals studied in this review could potentially serve as models for studying various human adrenal diseases. Each of them has its own characteristics and advantages, but also has certain limitations that may restrict their use in this role.
Among the species considered, spiny mice and golden hamster stand out for their morphological and functional similarity to the human adrenal glands. Their anatomical structure, including the arrangement of adrenal cortex zones and the expression of important enzymes for hormone synthesis, makes them the most promising model for research in the field of CAH.
Further molecular genetics and physiological studies of Acomys mice and golden hamsters may help establish their complete similarity to human adrenal glands and identify potential similarities and differences in pathologies. This could lead to the development of a “gold standard” animal model for studying human adrenal diseases, which in turn contributes to a more accurate understanding of the mechanisms of these diseases and the development of new treatment methods.
Data availability
Raw data used for the figure presented in this article will be made available upon request to the corresponding author.
Abbreviations
- 17β-HSD:
-
17β-hydroxysteroid dehydrogenase
- 3β-HSD:
-
3β-hydroxysteroid dehydrogenase
- ACTH:
-
adrenocorticotropic hormone
- CAH:
-
congenital adrenal hyperplasia
- CYT-B5:
-
cytochrome B5 enzyme
- DHEA:
-
dehydroepiandrosterone
- DHEA-S:
-
dehydroepiandrosterone sulphate
- IZ:
-
intermediate zone
- JZ:
-
juxtamedullary zone
- M:
-
medulla
- POR:
-
P450 oxidoreductase
- StAR:
-
acute steroidogenic regulatory protein
- SULT 2A1:
-
sulfotransferase
- ZF:
-
zona fasciculata
- ZG:
-
zona glomerulosa
- ZR:
-
zona reticularis
References
Miller WL, White PC. History of adrenal research: from ancient anatomy to contemporary molecular biology. Endocr Rev. 2023;44(1):70–116.
Navarro-Zambrana AN, Sheets LR. Ethnic and National Differences in Congenital Adrenal Hyperplasia Incidence: a systematic review and Meta-analysis. Horm Res Paediatr. 2023;96(3):249–58.
Kareva MA, Chugunov IS. Federal clinical practice guidelines on the management of the patients presenting with congenital adrenal hyperplasia. Probl Endocrinol. 2014;60(2):42–50. (In Russ.).
Buonocore F, Achermann JC. Primary adrenal insufficiency: new genetic causes and their long-term consequences. Clin Endocrinol (Oxf). 2020;92(1):11–20.
Fleming L, Van Riper M, Knafl K. Management of Childhood Congenital Adrenal Hyperplasia-An Integrative Review of the literature. J Pediatr Health Care. 2017;31:560–77.
Horrocks PM, London DR. Effects of long term dexamethasone treatment in adult patients with congenital adrenal hyperplasia. Clin Endocrinol (Oxf). 1987;27:635–42.
Young MC, Hughes IA. Dexamethasone treatment for congenital adrenal hyperplasia. Arch Dis Child. 1990;65:312–4.
Turcu AF, Auchus RJ. Novel treatment strategies in congenital adrenal hyperplasia. Curr Opin Endocrinol Diabetes Obes. 2016;23:225–32.
Glazova OV, Vorontsova MV, Sakr N, Shevkova LV, Onyanov NA, Kaziakhmedova SA, et al. Gene and cell therapy of adrenal pathology: achievements and prospects. Probl Endocrinol. 2021;67(6):80–9. (In Russ.).
Dörner M, Cochran J, Heikinheimo RS, Wilson M. Adrenocortical zonation, renewal, and remodeling. Front Endocrinol (Lausanne). 2015;6:27.
Mitani F. Functional zonation of the rat adrenal cortex: the development and maintenance. Proc Jpn Acad Ser B Phys Biol Sci. 2014;90(5):163–83.
Huang CC, Kang Y. The transient cortical zone in the adrenal gland: the mystery of the adrenal X-zone. J Endocrinol. 2019;241(1):51–63.
Yates R, Katugampola H, Cavlan D, Cogger K, Meimaridou E, Hughes C, et al. Adrenocortical development, maintenance, and disease. Curr Top Dev Biol. 2013;106:239–312.
Dagerlind A, Pelto-Huikko M, Diez M, Hökfelt T. Adrenal medullary ganglion neurons project into the splanchnic nerve. Neuroscience. 1995;69:1019–23.
Anderson DJ, Axel R. Molecular probes for the development and plasticity of neural crest derivatives. Cell. 1985;42:649–62.
Kameneva P, Artemov AV, Kastriti ME, Faure L, Olsen TK, Otte J, et al. Single-cell transcriptomics of human embryos identifies multiple sympathoblast lineages with potential implications for neuroblastoma origin. Nat Genet. 2021;53:694–706.
Wurtman RJ, Axelrod J. Adrenaline synthesis: control by the pituitary gland and adrenal glucocorticoids. Science. 1965;150:1464–5.
Ehrhart-Bornstein M, Bornstein SR. Cross-talk between adrenal medulla and adrenal cortex in stress. Ann N Y Acad Sci. 2008;1148:112–7.
Ross IL, Louw GJ. Embryological and molecular development of the adrenal glands. Clin Anat. 2015;28:235–42.
Jääskeläinen J. Molecular biology of androgen insensitivity. Mol Cell Endocrinol. 2012;352:4–12.
El Ghorayeb N, Bourdeau I, Lacroix A. Role of ACTH and other hormones in the regulation of Aldosterone production in primary Aldosteronism. Front Endocrinol (Lausanne). 2016;7:72.
Hyatt PJ, Bhatt K, Tait JF. Steroid biosynthesis by zona fasciculata and zona reticularis cells purified from the mammalian adrenal cortex. J Steroid Biochem. 1983;19:953–9.
Nadeau S, Rivest S. Glucocorticoids play a fundamental role in protecting the brain during innate immune response. J Neurosci. 2003;23:5536–44.
Kuo T, McQueen A, Chen TC, Wang JC. Regulation of glucose homeostasis by glucocorticoids. Adv Exp Med Biol. 2015;872:99–126.
Russell G, Lightman S. The human stress response. Nat Rev Endocrinol. 2019;15:525–34.
Hunter RW, Ivy JR, Bailey MA. Glucocorticoids and renal na + transport: implications for hypertension and salt sensitivity. J Physiol. 2014;592:1731–44.
Rege J, Nakamura Y, Satoh F, Morimoto R, Kennedy MR, Layman LC, et al. Liquid chromatography-tandem mass spectrometry analysis of human adrenal vein 19-carbon steroids before and after ACTH stimulation. J Clin Endocrinol Metab. 2013;98:1182–8.
Turcu AF, Auchus RJ. Adrenal steroidogenesis and congenital adrenal hyperplasia. Endocrinol Metab Clin North Am. 2015;44:275–96.
Nunes VS, Da Silva Ferreira G, Quintão ECR. Cholesterol metabolism in aging simultaneously altered in liver and nervous system. Aging. 2022;14:1549–61.
Miller WL, Geller DH, Rosen M. Ovarian and adrenal androgen biosynthesis and metabolism. Сontemporary Endocrinology. Humana Press. 2006;19–33.
Stocco DM. The role of the StAR protein in steroidogenesis: challenges for the future. J Endocrinol. 2000;164:247–53.
Slominski RM, Tuckey RC, Manna PR, Jetten AM, Postlethwaite A, Raman C, Slominski AT. Extra-adrenal glucocorticoid biosynthesis: implications for autoimmune and inflammatory disorders. Genes Immun. 2020;21:150–68.
Burger HG. Androgen production in women. Fertil Steril. 2002;77(4):3–5.
Mokrysheva NG, Melnichenko GA, Adamyan LV, Troshina EA, Molashenko NV, Sazonova AI, et al. Russian clinical practice guidelines «congenital adrenal hyperplasia. Obes Metabolism. 2021;18(3):345–82. (In Russ.).
Bakkar AA, Alsaedi A, Kamal NM, Althobaiti E, Aboulkhair LA, Almalki AM, et al. Lipoid congenital adrenal hyperplasia with a novel StAR gene mutation. Clin Med Insights Endocrinol Diabetes. 2023;16:11795514231167059.
Manna PR, Stetson CL, Slominski AT, Pruitt K. Role of the steroidogenic acute regulatory protein in health and disease. Endocrine. 2016;51(1):7–21.
Chien Y, Rosal K, Chung BC. Function of CYP11A1 in the mitochondria. Mol Cell Endocrinol. 2017;441:55–61.
Fujieda K, Okuhara K, Abe S, Tajima T, Mukai T, Nakae J. Molecular pathogenesis of lipoid adrenal hyperplasia and adrenal hypoplasia congenita. J Steroid Biochem Mol Biol. 2003;85:483–9.
Matteson KJ, Chung BC, Urdea MS, Miller WL. Study of cholesterol side-chain cleavage (20,22 desmolase) deficiency causing congenital lipoid adrenal hyperplasia using bovine-sequence P450scc oligodeoxyribonucleotide probes. Endocrinology. 1986;118:1296–305.
Picado-Leonard J, Miller WL. Cloning and sequence of the human gene for P450c17 (steroid 17 alpha-hydroxylase/17,20 lyase): similarity with the gene for P450c21. DNA. 1987;6:439–48.
Auchus RJ. The genetics, pathophysiology, and management of human deficiencies of P450c17. Endocrinol Metab Clin North Am. 2001;30:101–19.
Kater CE, Biglieri EG. Disorders of steroid 17 alpha-hydroxylase deficiency. Endocrinol Metab Clin North Am. 1994;23:341–57.
Biason-Lauber A, Leiberman E, Zachmann M. A single amino acid substitution in the putative redox partner-binding site of P450c17 as cause of isolated 17,20-lyase deficiency. J Clin Endocrinol Metab. 1997;82:3807–12.
Sherbet DP, Tiosano D, Kwist KM, Hochberg Z, Auchus RJ. CYP17 mutation E305G causes isolated 17,20-lyase deficiency by selectively altering substrate binding. J Biol Chem. 2003;278:48563–9.
Turkkahraman D, Guran T, Ivison H, Griffin A, Vijzelaar R, Krone N. Identification of a novel large CYP17A1 deletion by MLPA analysis in a family with classic 17α-hydroxylase deficiency. Sex Dev. 2015;9:91–7.
Brooke AM, Taylor NF, Shepherd JH, Gore ME, Ahmad T, Lin L, Rumsby G, Papari-Zareei M, Auchus RJ, Achermann JC, Monson JP. A novel point mutation in P450c17 (CYP17) causing combined 17alpha-hydroxylase/17,20-lyase deficiency. J Clin Endocrinol Metab. 2006;91:2428–31.
Van Den Akker EL, Koper JW, Boehmer AL, Themmen AP, Verhoef-Post M, Timmerman MA, Otten BJ, Drop SL, De Jong FH. Differential inhibition of 17alpha-hydroxylase and 17,20-lyase activities by three novel missense CYP17 mutations identified in patients with P450c17 deficiency. J Clin Endocrinol Metab. 2002;87:5714–21.
Al-Jurayyan NA. Ambiguous genitalia: two decades of experience. Ann Saudi Med. 2011;31:284–8.
Benkert AR, Young M, Robinson D, Hendrickson C, Lee PA, Strauss KA. Severe salt-losing 3β-Hydroxysteroid dehydrogenase Deficiency: treatment and outcomes of HSD3B2 c.35G > A homozygotes. J Clin Endocrinol Metab. 2015;100:1105–15.
Rhéaume E, Simard J, Morel Y, Mebarki F, Zachmann M, Forest MG, et al. Congenital adrenal hyperplasia due to point mutations in the type II 3 beta-hydroxysteroid dehydrogenase gene. Nat Genet. 1992;1:239–45.
Payne AH, Hales DB. Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr Rev. 2004;25:947–70.
Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, et al. Congenital adrenal Hyperplasia due to Steroid 21-Hydroxylase Deficiency: an endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2018;103:4043–88.
Concolino P, Costella A. Congenital adrenal Hyperplasia (CAH) due to 21-Hydroxylase Deficiency: a Comprehensive Focus on 233 pathogenic variants of CYP21A2 gene. Mol Diagn Ther. 2018;22:261–80.
Riedl S, Röhl FW, Bonfig W, Brämswig J, Richter-Unruh A, Fricke-Otto S, et al. Genotype/phenotype correlations in 538 congenital adrenal hyperplasia patients from Germany and Austria: discordances in milder genotypes and in screened versus prescreening patients. Endocr Connect. 2019;8:86–94.
Pallan PS, Wang C, Lei L, Yoshimoto FK, Auchus RJ, Waterman MR, et al. Human cytochrome P450 21A2, the major steroid 21-Hydroxylase: structure of the enzyme·progesterone substrate complex and rate-limiting c-h bond cleavage. J Biol Chem. 2015;290:13128–43.
Neunzig J, Milhim M, Schiffer L, Khatri Y, Zapp J, Sánchez-Guijo A, et al. The steroid metabolite 16(β)-OH-androstenedione generated by CYP21A2 serves as a substrate for CYP19A1. J Steroid Biochem Mol Biol. 2017;167:182–91.
Guengerich FP, Waterman MR, Egli M. Recent structural insights into cytochrome P450 function. Trends Pharmacol Sci. 2016;37:625–40.
Sushko TA, Gilep AA, Usanov SA. Mechanism of intermolecular interactions of microsomal cytochrome P450s CYP17 and CYP21 involved in steroid hormone biosynthesis. Biochem (Mosc). 2012;77:585–92.
Kohn B, Levine LS, Pollack MS, Pang S, Lorenzen F, Levy D, et al. Late-onset steroid 21-hydroxylase deficiency: a variant of classical congenital adrenal hyperplasia. J Clin Endocrinol Metab. 1982;55:817–27.
Tusie-Luna MT, Traktman P, White PC. Determination of functional effects of mutations in the steroid 21-hydroxylase gene (CYP21) using recombinant vaccinia virus. J Biol Chem. 1990;265:20916–22.
White PC. Steroid 11 beta-hydroxylase deficiency and related disorders. Endocrinol Metab Clin North Am. 2001;30:61–79.
Nimkarn S, New MI. Steroid 11beta- hydroxylase deficiency congenital adrenal hyperplasia. Trends Endocrinol Metab. 2008;19:96–9.
Krone N, Arlt W. Genetics of congenital adrenal hyperplasia. Best Pract Res Clin Endocrinol Metab. 2009;23:181–92.
Asla Q, Sardà H, Lerma E, Hanzu FA, Rodrigo MT, Urgell E, et al. 11-Deoxycorticosterone producing adrenal Hyperplasia as a very unusual cause of endocrine hypertension: case report and systematic review of the literature. Front Endocrinol (Lausanne). 2022;13:846865.
Adachi M, Tachibana K, Asakura Y, Yamamoto T, Hanaki K, Oka A. Compound heterozygous mutations of cytochrome P450 oxidoreductase gene (POR) in two patients with Antley-Bixler syndrome. Am J Med Genet A. 2004;128A:333–9.
Pandey AV, Flück CE. NADPH P450 oxidoreductase: structure, function, and pathology of diseases. Pharmacol Ther. 2013;138:229–54.
Flück CE, Pandey AV, Huang N, Agrawal V, Miller WL. P450 oxidoreductase deficiency - a new form of congenital adrenal hyperplasia. Endocr Dev. 2008;13:67–81.
Owens SL, Downey ME, Pressler BM, et al. Congenital adrenal hyperplasia associated with mutation in an 11β-hydroxylase-like gene in a cat. J Vet Intern Med. 2012 Sep-Oct;26(5):1221–6.
Holmes RL. The adrenal glands of the ferret, Mustela putorius. J Anat. 1961;95:325–36.
Bakthavatchalu V, Muthupalani S, Marini RP, Fox JG. Endocrinopathy and Aging in ferrets. Vet Pathol. 2016;53:349–65.
Auchus RJ. Overview of dehydroepiandrosterone biosynthesis. Semin Reprod Med. 2004;22:281–8.
Wagner S, Kiupel M, Peterson RA, Heikinheimo M, Wilson DB. Cytochrome b5 expression in gonadectomy-induced adrenocortical neoplasms of the domestic ferret (Mustela putorius furo). Vet Pathol. 2008;45:439–42.
Bielinska M, Kiiveri S, Parviainen H, Mannisto S, Heikinheimo M, Wilson DB. Gonadectomy-induced adrenocortical neoplasia in the domestic ferret (Mustela putorius furo) and laboratory mouse. Vet Pathol. 2006;43:97–117.
Kaneko JJ, Harvey JW, Bruss ML. Clinical biochemistry of domestic animals. Academic; 2008.
Rosenthal KL, Peterson ME. Evaluation of plasma androgen and estrogen concentrations in ferrets with hyperadrenocorticism. J Am Vet Med Assoc. 1996;209:1097–102.
Marini RP, Otto G, Erdman S, Palley L, Fox JG. Biology and diseases of ferrets. Lab Anim Med. 2002:483–517.
Mabry J, Rowe T, Blanchard E, Xu X. Development of a HEPA-filtered caging system for ferrets: thinking outside the metal box. Lab Anim Sci Prof. 2013;1:41–4.
Oh DY, Hurt AC. Using the Ferret as an animal model for investigating influenza antiviral effectiveness. Front Microbiol. 2016;7:80.
Kempná P, Flück CE. Adrenal gland development and defects. Best Pract Res Clin Endocrinol Metab. 2008;22:77–93.
Gomez-Sanchez CE, Ferris MW, Foecking MF, Gomez-Sanchez EP. Synthesis of 18-hydroxycortisol and 18-oxocortisol in bovine adrenal slices. J Steroid Biochem. 1989;33:595–8.
Ramsey IK. Trilostane in dogs. Vet Clin North Am Small Anim Pract. 2010;40:269–83.
Lisurek M, Bernhardt R. Modulation of aldosterone and cortisol synthesis on the molecular level. Mol Cell Endocrinol. 2004;215:149–59.
Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev. 2011;32(1):81–151. Erratum in: Endocr Rev. 201;4:579.
Sanders K, Mol JA, Kooistra HS, Slob A, Galac S. New insights in the functional zonation of the canine adrenal cortex. J Vet Intern Med. 2016;30:741–50.
Frank LA, Rohrbach BW, Bailey EM, West JR, Oliver JW. Steroid hormone concentration profiles in healthy intact and neutered dogs before and after cosyntropin administration. Domest Anim Endocrinol. 2003;24:43–57.
Beuschlein F, Galac S, Wilson DB. Animal models of adrenocortical tumorigenesis. Mol Cell Endocrinol. 2012;351:78–86.
Altman D. Starting With Guinea Pigs. 1999.
Rowlands IW, Weir BJ. The biology of hystricomorph rodents. Symp Zool Soc Lond UK. 1974;34.
Lafi A, Mirhish S. Histomorphological and Histochemical Study of Adrenal Gland in Adult Male of Guinea pigs (Cavia porcellus). Iraqi J Veterinary Med. 2019;43:59–66.
Banks WJ. Applied Veterinary Histology. Endocrine system 3rd ed. Mosby Year Book Baltim Boston Lond. 1993:408–27.
Sheikhian A, Saadatfar Z, Mohammadpour AA. A histological study of adrenal gland in guinea pig and hamster. Comp Clin Pathol. 2015;24:1069–74.
Le Goascogne C, Sananès N, Gouézou M, Takemori S, Kominami S, Baulieu EE, et al. Immunoreactive cytochrome P-450(17 alpha) in rat and guinea-pig gonads, adrenal glands, and brain. J Reprod Fertil. 1991;93:609–22.
Rystrom TL, Prawitt RC, Richter SH, Sachser N, Kaiser S. Repeatability of endocrine traits and dominance rank in female guinea pigs. Front Zool. 2022;19:4.
Bélanger B, Caron S, Boudou P, Fiet J, Bélanger A. Adrenal steroidogenesis in the guinea pig: effects of androgens. Steroids. 1992;57:76–81.
Bélanger B, Caron S, Bélanger A, Dupont A. Steroid fatty acid esters in adrenals and plasma: effects of ACTH. J Endocrinol. 1990;127:505–11.
Provencher P, Lorrain A, Bélanger A, Fiet J. Steroid biosynthesis by zona glomerulosa-fasciculata cells in primary culture of guinea-pig adrenals. J Steroid Biochem. 1990;36:589–96.
Provencher PH, Tremblay Y, Caron S, Belanger A. Effect of chronic ACTH treatment on guinea-pig adrenal steroidogenesis: steroid plasma levels, steroid adrenal levels, activity of steroidogenic enzymes and their steady-state mRNA levels. J Steroid Biochem Mol Biol. 1992;41:69–78.
Altman DietrichWarner BM, Safronetz D, Kobinger GP. Syrian hamsters as a small animal model for emerging infectious diseases: advances in immunologic methods. Adv Exp Med Biol. 2017;972:87–101.
Peczenik O. Actions of sex hormones on ≈Уstrous cycle and reproduction of the Golden Hamster. Journ Endocrin. 1942;3:157–67.
Morton A. Observations on the histophysiology of the adrenal gland of the golden hamster. Endocrinology. 1950;46:166–76.
Holmes WN. Histological variations in the adrenal cortex of the golden hamster with special reference to the X zone. Anat Rec. 1955;122:271–93.
Keyes PH. Adreno-cortical changes in Syrian hamsters following gonadectomy. Endocrinology. 1949;44:274–7.
Koneff AA, Simpson ME, Evans HM. Effects of chronic administration of diethylstilbestrol on the pituitary and other endocrine organs of hamsters. Anat Rec. 1946;94:169–95.
Cloutier M, Fleury A, Courtemanche J, Ducharme L, Mason JI, Lehoux JG. Characterization of the adrenal cytochrome P450C17 in the hamster, a small animal model for the study of adrenal dehydroepiandrosterone biosynthesis. DNA Cell Biol. 1997;16:357–68.
Briére N, Martel D, Cloutier M, LeHoux JG. Immunolocalization and biochemical determination of cytochrome P450C17 in adrenals of hamsters treated with ACTH. J Histochem Cytochem. 1997;45:1409–16.
Rogerson FM, LeHoux JG, Mason JI. Expression and characterization of isoforms of 3 beta-hydroxysteroid dehydrogenase/delta 5–>4-isomerase in the hamster. J Steroid Biochem Mol Biol. 1995;55:481–7.
Suzuki T, Kachi T. Similarities and differences in supporting and chromaffin cells in the mammalian adrenal medullae: an immunohistochemical study. Anat Rec. 1996;244:358–65.
Niepoth N, Merritt JR, Uminski M et al. Evolution of a novel adrenal cell type that promotes parental care. Nature. 2024.
Perleberg C, Kind A, Schnieke A. Genetically engineered pigs as models for human disease. Dis Model Mech. 2018;11:dmm030783.
Renner S, Blutke A, Clauss S, Deeg CA, Kemter E, Merkus D, et al. Porcine models for studying complications and organ crosstalk in diabetes mellitus. Cell Tissue Res. 2020;380:341–78.
Moretti A, Fonteyne L, Giesert F, Hoppmann P, Meier AB, Bozoglu T, et al. Somatic gene editing ameliorates skeletal and cardiac muscle failure in pig and human models of Duchenne muscular dystrophy. Nat Med. 2020;26:207–14.
Renner S, Römisch-Margl W, Prehn C, Krebs S, Adamski J, Görke B, et al. Changing metabolic signatures of amino acids and lipids during the prediabetic period in a pig model with impaired incretin function and reduced β-cell mass. Diabetes. 2012;61:2166–75.
Streckel E, Braun-Reichhart C, Herbach N, Dahlhoff M, Kessler B, Blutke A, et al. Effects of the glucagon-like peptide-1 receptor agonist liraglutide in juvenile transgenic pigs modeling a pre-diabetic condition. J Transl Med. 2015;13:73.
Kumar V, Sharma A. Gross and histological observations on adrenal gland of Pig. Indian J Vet Anat. 2019;31:117–8.
Vohra T, Kemter E, Sun N, Dobenecker B, Hinrichs A, Burrello J, et al. Effect of Dietary Sodium Modulation on Pig adrenal steroidogenesis and transcriptome profiles. Hypertension. 2020;76:1769–77.
Robic A, Faraut T, Prunier A. Pathways and genes involved in steroid hormone metabolism in male pigs: a review and update. J Steroid Biochem Mol Biol. 2014;140:44–55.
Lowenstine LJ. A primer of primate pathology: lesions and nonlesions. Toxicol Pathol. 2003 Jan-Feb;31 Suppl:92–102. doi: 10.1080/01926230390177668. PMID: 12597436.
Tachibana T, Kusakabe KT, Osaki S, Kuraishi T, Hattori S, Yoshizawa M, Kai C, Kiso Y. Histocytological specificities of adrenal cortex in the New World monkeys, Aotus lemurinus and Saimiri boliviensis. J Vet Med Sci. 2016;78(1):161–5.
Parker CR Jr, Jian M, Conley AJ. The localization of DHEA sulfotransferase in steroidogenic and steroid metabolizing tissues of the adult rhesus macaque monkey. Endocr Res. 2000;26(4):517–22.
Mapes S, Corbin CJ, Tarantal A, Conley A. The primate adrenal zona reticularis is defined by expression of cytochrome b5, 17alpha-hydroxylase/17,20-lyase cytochrome P450 (P450c17) and NADPH-cytochrome P450 reductase (reductase) but not 3beta-hydroxysteroid dehydrogenase/delta5-4 isomerase (3beta-HSD). J Clin Endocrinol Metab. 1999;84(9):3382–5.
Pattison JC, Abbott DH, Saltzman W, Nguyen AD, Henderson G, Jing H, Pryce CR, Allen AJ, Conley AJ, Bird IM. Male marmoset monkeys express an adrenal fetal zone at birth, but not a zona reticularis in adulthood. Endocrinology. 2005;146(1):365–74.
Streeter KA, Sunshine MD, Brant JO, Sandoval AGW, Maden M, Fuller DD. Molecular and histologic outcomes following spinal cord injury in spiny mice, Acomys cahirinus. J Comp Neurol. 2020;528:1535–47.
Chevret P, Denys C, Jaeger JJ, Michaux J, Catzeflis FM. Molecular evidence that the spiny mouse (Acomys) is more closely related to gerbils (Gerbillinae) than to true mice (Murinae). Proc Natl Acad Sci U S A. 1993;90:3433–6.
Aghová T, Palupčíková K, Šumbera R, Frynta D, Lavrenchenko LA, Meheretu Y, et al. Multiple radiations of spiny mice (Rodentia: Acomys) in dry open habitats of Afro-Arabia: evidence from a multi-locus phylogeny. BMC Evol Biol. 2019;19:69.
Lamers WH, Mooren PG, Griep H, Endert E, Degenhart HJ, Charles R. Hormones in perinatal rat and spiny mouse: relation to altricial and precocial timing of birth. Am J Physiol. 1986;251:E78–85.
Seifert AW, Kiama SG, Seifert MG, Goheen JR, Palmer TM, Maden M. Skin shedding and tissue regeneration in African spiny mice (Acomys). Nature. 2012;489:561–5.
Bonhote J. Exhibition of and remarks upon a young Cairo Spiny Mouse (Acomys cahirinus). Proc Zool Soc Lond. 1911;5:5–6.
Creutzfeldt W, Mende D, Willms B, Söling HD. Vascular basement membrane thickness in muscle of spiny mice and activities of glycolysis and gluconeogenesis in the liver of animals with spontaneous and experimental diabetes and of untreated human diabetics. Diabetologia. 1970;6:356–60.
Shkolnik A, Borut A. Temperature and water relations in two species of spiny mice (Acomys). J Mammal. 1969;50:245–55.
Bellofiore N, Ellery SJ, Mamrot J, Walker DW, Temple-Smith P, Dickinson H. First evidence of a menstruating rodent: the spiny mouse (Acomys cahirinus). Am J Obstet Gynecol. 2017;216:e401–4011.
Quinn TA, Ratnayake U, Dickinson H, Nguyen TH, McIntosh M, Castillo-Melendez M, et al. Ontogeny of the adrenal gland in the spiny mouse, with particular reference to production of the steroids cortisol and dehydroepiandrosterone. Endocrinology. 2013;154:1190–201.
Quinn TA, Ratnayake U, Castillo-Melendez M, Moritz KM, Dickinson H, Walker DW. Adrenal steroidogenesis following prenatal dexamethasone exposure in the spiny mouse. J Endocrinol. 2014;221:347–62.
Acknowledgements
Not applicable.
Funding
Supported by Grant 075-15-2021-1344 of MSER and 075-15-2022-310 by MSER.
Author information
Authors and Affiliations
Contributions
Conceptualization by Al.B., A.B. and O.G; writing—original draft preparation by Al.B., A.B., N.F.; writing—review and editing by M.V. and O.G. supervision by E.Sh., A.K and O.G. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bilyalova, A., Bilyalov, A., Filatov, N. et al. Non-classical animal models for studying adrenal diseases: advantages, limitations, and implications for research. Lab Anim Res 40, 25 (2024). https://doi.org/10.1186/s42826-024-00212-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42826-024-00212-8