
Abstract
In the present work, a comparative study of analytical methods for the simultaneous and quantitative determination of trivalent and hexavalent chromium is presented. For the analysis by ion chromatography-inductively coupled plasma-mass spectrometry, two different columns were tested, as well as different mobile phases and different pH of the samples. The optimized analytical method permitted the separation of Cr(III) and Cr(VI) using 75 mmol/L NH4NO3 pH 3 as chromatographic eluent. The method was validated and applied to real samples, allowing the determination of both species simultaneously, even when there is a huge difference of concentration between Cr(III) and Cr(VI). Limit of detection and limit of quantification for Cr(III) were found to be 0.016 and 0.054 \(\upmu\)g/L (0.3 and 1.1 \(\upmu\)g/kg), respectively, and for Cr(VI) 0.13 and 0.43 \(\upmu\)g/L (7 and 22 \(\upmu\)g/kg), respectively. Possible species interconversions were monitored through the use of chromium isotopic standards, which confirmed that the optimized methodology preserves chromium speciation during extraction and analysis. Fourier-transform ion cyclotron resonance-mass spectrometry permitted the structure elucidation of the complex formed during ethylenediaminetetraacetic acid extraction.
Graphical Abstract

Similar content being viewed by others

1 Introduction
Tanning is the process of transforming raw hide into leather. Among the different tanning methods, chrome tanning is the most common technique and accounts for around 90% of global leather production [1]. Chrome tanning uses trivalent chromium salts as tanning agents [2]. The multiple advantages of this process (fast tanning reaction, it confers high hydrothermal stability, it allows the retention of colorants, etc. [3, 4]) make chrome tanning the most widespread technique.
Trivalent chromium (Cr(III)) is not considered as toxic but it can be unintentionally oxidized into hexavalent chromium (Cr(VI)), a known mutagenic and carcinogenic [5], as a consequence of different factors [6, 7]. Additionally, in direct contact with the skin, chrome-tanned leather products may elicit allergic contact dermatitis [8, 9]. The European Union regulates hexavalent chromium’s content to 3 mg/kg in leather goods coming into direct contact with skin [10], while the leather standard by Oeko-Tex® limits total chromium content to 2 mg/kg for leather products intended for babies [11].
Concerning hexavalent chromium determination, the International Organization for Standardization (ISO) 17075 standard describes a procedure for the selective extraction of Cr(VI) in tanned leather using a phosphate buffer: K2HPO4·3H2O at pH 8.0. The analysis of the extract is performed via colorimetric [12] or chromatographic analysis [13]. Both methods rely on the complexation of Cr(VI) with 1,5-diphenylcarbazide (DPC) for UV–Vis detection and offer a limit of quantification (LOQ) of 3 mg/kg for Cr(VI). Some other authors have reported the determination of Cr(VI) in leather samples based on its extraction with a phosphate buffer [14, 15], NaOH [16] or through a sequential extractions scheme [1]. However, the determination still relies on the complexation of Cr(VI) with DPC, and whilst this technique has proven to be effective, an inconvenient may surge due to the interference of colorants that may absorb at the same wavelength (540 nm) as the complex formed between Cr(VI) and DPC. On the other side, trivalent chromium has been generally measured by subtracting Cr(VI) from the total Cr content [5]. However, recent studies have revealed that from an allergic and exposure perspective, Cr(III) is also relevant [9], this is why it is important to quantify both chromium species separately [17].
Several articles have been published regarding chromium speciation in other matrices such as foodstuffs [18,19,20], cosmetic products [21], environmental [22,23,24], and pharmaceutical samples [25, 26] by means of hyphenation techniques such as ion chromatography-inductively coupled plasma-mass spectrometry (IC-ICP-MS), liquid chromatography-inductively coupled plasma-mass spectrometry, ion chromatography—tandem mass spectrometry, etc., but to our knowledge, the simultaneous extraction Cr(VI) and Cr(III), while maintaining the speciation, has not been studied for leather products. The aim of this work was to develop an analytical method to quantitatively and simultaneously determine Cr(VI) and Cr(III) as a chromium-EDTA complex in leather samples in the same run, limiting the interconversions during extraction and analysis, which were followed via chromium isotopic standards. While the other reported methods for leather samples are focused on Cr(VI), in this study the simultaneous dosage of both chromium species is developed. A second challenge was to deal with a high amount of Cr(III), compared to Cr(VI), in real leather samples. In order to achieve all of these objectives, different conditions were tested for the analysis through IC-ICP-MS, which allows to have information on the isotope and also in which ionic form the isotope is present. Some other advantages of this hyphenation are its high sensitivity, high selectivity and low detection limits. For example, while the ISO 17075 protocol offers a LOQ of 3 mg/kg for Cr(VI) through ion chromatography-UV–visible detection, IC-ICP-MS offers a LOQ of 0.6–0.8 µg/kg for Cr(VI) [18] in dairy and cereal products, and 38 µg/kg for Cr(III) [19] in dairy products.
2 Experimental
2.1 Instrumentation
An ETHOS One microwave digestion system (Milestone, Sorisole, Italy) equipped with a temperature sensor was used to perform the extractions and complexations. TFM reactors were employed with a maximum pressure of 100 bar. The pH of the solutions was determined with a PHM240 pH/Ion Meter (MeterLab, Radiometer Analytical, Villeurbanne, France). The extracts were centrifuged with a 5702 Centrifuge (Eppendorf, Montesson, France).
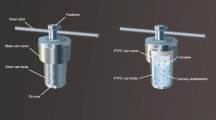
A Dionex ICS-6000 HPIC System (Thermo Fisher Scientific, Courtaboeuf, France) permitted species separation. For the optimized conditions, the ion chromatography pump delivered the mobile phase and the sample at 0.25 mL/min. A Dionex ADRS 600 Anion Dynamically Regenerated Suppressor (2 mm) (Thermo Fisher Scientific) permitted to avoid salt deposition and was set before the entrance into the Inductively Coupled Plasma—Mass Spectrometer (ICP-MS). The suppressor was operated in external regeneration mode achieved by the supply of ultrapure water at a constant flow of 0.6 mL/min through the use of a Dionex AXP auxiliary pump (Thermo Fisher Scientific). Columns were placed in the following order: a Dionex IonPac NG1 guard column (2 × 50 mm, particle size: 10 µm), a Dionex IonPac AG7 guard column (2 × 50 mm, particle size: 10 µm) and a Dionex IonPac AS7 analytical column (2 × 250 mm, particle size: 10 µm). A PEEK tubing (0.25 mm i.d., 150 cm long) permitted to couple the analytical column to the nebulizer of the ICP-MS. Finally, the detection of Cr species was performed on an iCAP RQ ICP-MS instrument (Thermo Fisher Scientific, Courtaboeuf, France) fitted with a collision cell. Two different measurement modes were tested: standard mode (STD) and kinetic energy discrimination mode (KED). In the KED mode, a non-reactive gas (helium) was injected in the collision cell to selectively remove unwanted polyatomic interferences. The Qtegra software (Thermo Fisher Scientific) permitted to process the data obtained. The optimized conditions for the plasma and nebulizer for ICP-MS are described in Table 1. Figure 1a shows a diagram of the setup used for the optimized conditions. For each setup (optimized conditions and tests), mobile phases were previously degassed using an Elmasonic S ultrasonic bath (Elma, Singen, Germany).

Chromatographic setups. a For the optimized separation conditions (AS7 column, NH4NO3 as eluent in isocratic mode). b For the CS5A column using HNO3 as eluent in gradient mode
A second setup was tested when trying to optimize the separation conditions (Fig. 1b). For these experiments, a Dionex IonPac NG1 guard column (2 × 50 mm, particle size: 10 µm), a Dionex IonPac CG5A guard column (2 × 50 mm, particle size: 9 µm) and a Dionex IonPac CS5A ion-exchange column (2 × 250 mm, particle size: 9 µm) were installed. In the same way as the first setup, the ion chromatography pump delivered the mobile phase (A: HNO3 1.1 M, B: ultrapure water) and the sample 0.275 mL/min, but in this case, in gradient mode (0–32% A: 60 s; 32% A: 102 s; 100% A: 360 s; 0% A: 1200 s). This solution traveled through the columns where species were separated and eluted at different retention times. After passing through the conductivity detector, a Y-connector was installed. This permitted the arrival of the sample and the mobile phase at 0.275 mL/min from one side, and the arrival of ultrapure water at 0.8 mL/min by means of an AXP auxiliary pump (Thermo Fisher Scientific) from the other side. The final solution (at the exit of the Y-connector) was diluted nitric acid. This solution finally arrived to the ICP-MS where detection was completed.
Preliminary tests were conducted using a NG1 guard column and an AS7 ion-exchange column with nitric acid as mobile phase (no suppressor, no AXP pump neither). FTICR MS analyses were achieved using a SolariX XR instrument (Bruker Daltonics, Bremen, Germany) equipped with a 12 T superconducting magnet and a dynamically harmonized ICR cell (ParaCell®). An ESI source was used in positive and negative ion modes. The operating parameters for direct infusion analyses on ESI(+) and ESI(−) are reported in Table 2. The analogue image current was digitized with 4 million data points resulting in the recording of a 1.118 s time domain signal for ESI analyses. The time domain signals were transformed into the corresponding frequency domain by Fourier transform (after full-sine apodization and one zero fill). These settings resulted in a mass resolving power of 500,000 at m/z 200. Mass spectra were acquired using Bruker Compass FtmsControl 2.3, and processed with Bruker Compass DataAnalysis 5.1 (Bruker Daltonics GmbH, Bremen, Germany). Molecular formulas were assigned to the peaks with a signal-to-noise ratio greater than 6, with even and odd electron configuration.
2.2 Materials
Ultrapure water (resistivity 18.2 MW cm), obtained from a Milli-Q system (Millipore, Bedford, MA, USA), was used for preparation of all solutions unless stated otherwise. Nitric acid 67–69% (Ultrapure Normatom, VWR Chemicals, Leuven, Belgium) and NH4NO3 (Normapur guaranteed reagent, Prolabo, Paris, France) were used for the preparation of mobile phases. Chromium standards were prepared with chromium(III) nitrate nonahydrate (99%, Thermo Scientific, Geel, Belgium) and potassium dichromate (R.P. Normapur, Prolabo, Paris, France). For the extractions and complexations, K2HPO4∙3H2O (AnalaR Normapur, VWR Chemicals, Leuven, Belgium), phosphoric acid (85 wt.% in H2O, Sigma-Aldrich, Germany), EDTA (Analytical reagent, VWR Chemicals, Leuven, Belgium) and NH4OH (20–22% NH3) (Optima grade, Fisher Scientific, UK) were employed. Filtration was performed with 0.45 µm nylon syringe filters (Fisher Scientific). Leather samples came from the luxury industry and were all chrome-tanned and colored. Different types of leather were used for the extractions, such as: chrome-tanned, colored, polished and satin leather. 53Cr(III) (97.01%) and 50Cr(VI) (97.36%) chromium isotopic standards (100 ppm, Certified Reference Material, ISC Science, Oviedo, Spain) were diluted and used to monitor possible chromium interconversions. Chromium(III) nitrate nonahydrate was used for preparing the chromium-EDTA complex analyzed by Fourier-transform ion cyclotron resonance-mass spectrometry (FTICR-MS). Optima LC–MS grade water and acetonitrile (purchased from Fisher Chemical) were employed for the dilution of samples analyzed by FTICR-MS.
2.3 Methodology
EDTA extractions/complexations were carried out following the procedures reported in a previous study [17]: 200 mg of leather were extracted with 4 mL of EDTA 50 mmol/L (adjusted to pH 10 with NH4OH) at 100 °C in a microwave digestion system. On the other hand, phosphate buffer extractions were performed with K2HPO4·3H2O at pH 8.0 for 3 h under magnetic stirring, and the extracts were subsequently filtrated through nylon 0.45 µm filter, as described in ISO 17075 protocol [12]. In order to follow possible interconversions, chromium isotopic standards were used instead of the leather samples and followed the whole ISO 17075 and EDTA extraction protocols.
Regarding chromium speciation by IC-ICP-MS, preliminary studies were performed using nitric acid as mobile phase with the setup previously described in Sect. 2.1. For the chromatographic setup used for the CS5A column (Fig. 1b), HNO3 1.1 M was used as mobile phase in gradient mode. Optimized conditions were found when using the AS7 ion-exchange column. For the optimized setup (Fig. 1a), 75 mmol/L NH4NO3 adjusted to pH 3 with nitric acid 1 M was used as chromatographic eluent.
As for FTICR-MS analyses, samples were diluted as follows: 2X dilution in acetonitrile for chromium-EDTA complexes [initial concentration of 10 ppm of Cr(III)] and 2X dilution in acetonitrile/H2O (50/50, v/v) for non-diluted leather extracts. Subsequently, solutions were injected and analyzed in the conditions previously indicated. Mass spectra were internally calibrated using confident ions signal over the whole mass range.
3 Results and discussion
3.1 Speciation by IC-ICP-MS
3.1.1 Preliminary tests
Preliminary speciation studies were performed with nitric acid as mobile phase as described on the application note by Thermo Scientific [27] for the speciation analysis of Cr(III) and Cr(VI) in drinking waters. However, the protocol required 0.4 mol/L (approximately 2.5% v/v) nitric acid, and the ICP-MS should not be used at acidic concentrations beyond 2% (v/v). Therefore, HNO3 1.9% (v/v) was used instead to prevent corrosion of the cones. Additionally, an AS7 ion-exchange column replaced the AG7 guard column proposed by the protocol. Some of the advantages of using nitric acid as mobile phase are the fact that it allows the separation of Cr(III) and Cr(VI) while preserving speciation, and the easy implementation of the methodology since no suppressor (and thus no AXP pump) is required in view of the fact that nitric acid is compatible with the ICP-MS system. The use of other mobile phases such as ammonium nitrate requires de implementation of a different setup in order to allow salt deposition (suppression of the mobile phase, an additional pump to supply water for suppressor regeneration).
One of the first parameters to optimize when starting to work with the IC-ICP-MS hyphenation was the dwell time of the mass spectrometer, which is the period of time that the instrument accumulates the signal or the time spent for acquiring data. Dwell times tested were: 0.02, 0.05, 0.07, 0.2, 0.5, and 1 s. From all of these values, 0.05 s provided the best results in terms of chromatographic signal-to-noise ratio. Consequently, this was the value used for all the IC-ICP-MS analyses hereafter.
Once the dwell time was optimized, several conditions were tested with the aim of studying the retention of hexavalent and trivalent chromium species, such as concentration of the mobile phase, flow rate, temperature of the column compartment, and the addition of organic solvent to the mobile phase. Regarding this last parameter, it was noticed that the addition of 5% acetonitrile (ACN) at a flow rate of 0.6 mL/min decreased the retention time of Cr(III) from 940 to 860 s and the retention time of Cr(VI) from 130 to 125 s as compared to the condition without organic solvent (HNO3 1.9% (v/v), 0.6 mL/min). According to Rabin and Stillian [28], acetonitrile causes a swelling of the latex containing the ion-exchange sites which provokes a less effective cross-linking of the latex and decreases the number of sites per unit area, hence decreasing the time that ions spend bound to the column.
Moreover, on a previous study [17] EDTA proved to be the most performant method to extract Cr(VI) and Cr(III) as Cr(III)-EDTA− from leather samples, hence the retention of Cr(III)-EDTA− was studied as well.
Table 3 shows the retention times of the different peaks observed after injecting Cr(III)-EDTA− samples at different pH using a NG1 guard column followed by an AG7 guard column for species separation. HNO3 1.9% (v/v) was used as mobile phase at a flow rate of 0.3 mL/min. The objective of this test was to study the stability of the complex at basic and acidic medium.
For the first injection, the sample had a pH of 11 which was achieved by adjusting the pH of the sample with a NaOH solution. The second sample was injected at a pH of 6, accomplished by merely diluting the sample in water. A Cr(III)-EDTA− peak was observed but also Cr(VI) peak, which proved that some Cr(III) were transformed into Cr(VI). Finally, the third sample was injected at pH 2 which was achieved by acidifying the sample with nitric acid (final solution acidic concentration: HNO3 0.1% v/v). It could be concluded from this part that acidifying the samples maintained the stability of Cr(III)-EDTA− and no decomplexation was observed (only one peak was observed at 55 s). However, the complex was not retained enough to consider this method as valuable.
After preliminary studies, different columns and mobile phases were explored. A nitric acid solution as well as an ammonium nitrate solution were tested as mobile phases. For each mobile phase, two different columns (and thus chromatographic setups) were tested: an AS7 anion-exchange column and a CS5A ion-exchange column. A summary of the different conditions that were tested can be seen in Supplementary Information (Additional file 1: Figure S1).
3.1.2 AS7 column and nitric acid as mobile phase
After testing several conditions during preliminary tests and considering that Cr(III)-EDTA− elutes very close to the dead time, the retained conditions for this configuration were: NG1 guard column + AS7 column, mobile phase: HNO3 1.9% (v/v), and flow rate: 0.4 mL/min. Samples were injected with an HNO3 concentration of 0.1% (v/v) in order to preserve the stability of the complex.
This permitted to separate Cr(III) and Cr(VI). Cr(VI) eluted at 190 s, while Cr(III) eluted at 1300 s. Nevertheless, Cr(III)-EDTA− eluted at 75 s, still very close to the dead time which was 70 s. This can be appreciated in the chromatograms shown in Fig. 2. In fact, the AS7 ion-exchange column is a latex-based pellicular column. Its central polymeric packing is sulfonated (cation exchanger) while the latex carries quaternary ammonium groups (anion exchanger). Therefore, this column contains both anionic and cationic ion-exchange capacity [29]. The latex is electrostatically agglomerated to the central backbone. The particularity of this structure induces a possible penetration of metallic cations (whose hydration sphere is reduced) towards the sulfonates of the central bead, and thus, their retention. In classical analysis (eluent of lower ionic strength and basic pH) there would be a more important retention of anionic species (Cr(VI) species) and the metal cation (Cr(III)) would remain stuck to the core of the stationary phase. The use of acid allows the elution of Cr(III) since the ionic strength of the nitrate at acidic pH has the purpose of minimizing the retention of anions. Being Cr(III)-EDTA− a big molecular structure compared to the size of the pores of the stationary phase, it has little entrance into the pores and flows freely. This explains why the complex elutes at first (retention time = 75 s) and very close to the dead time, followed by Cr(VI) which elutes 115 s later (retention time = 190 s). Cr(III) elutes at last, when nitric acid succeeds in removing the cation from the core of the central bead.

IC-ICP-MS chromatograms obtained through the AS7 column using nitric acid as eluent. a Cr(VI) and Cr(III), 50 ppb, HNO3 0.1% (v/v). b 50 ppb of Cr(III) in presence of EDTA (after going through the EDTA extraction/complexation conditions), HNO3 0.1% (v/v). NG1 guard column + AS7 column, mobile phase: HNO3 1.9% (v/v), flow rate: 0.4 mL/min, STD mode, dead time: 70 s
3.1.3 CS5A column and nitric acid as mobile phase
The use of a CS5A column using nitric acid as eluent was tested based on the work by Hernandez et al., who reported a methodology for the determination of Cr(III) and Cr(VI) in dairy products through high performance liquid chromatography—inductively coupled plasma—mass spectrometry (HPLC-ICP-MS). They proposed two different gradient programs for the determination of Cr(III) [19] and Cr(VI) [18], separately, as well as two different extraction methods. Nonetheless, in order to adapt the gradient program to the current chromatographic conditions, the HPLC gradient method transfer calculator from University of Geneva was used [30, 31]. The method calculated was later optimized in order to be able to use one sole gradient program to elute both chromium species, Cr(III) and Cr(VI), together with Cr(III)-EDTA−. The final gradient is described in Table 4. The setup used for these tests is depicted in Fig. 1b.
The EDTA extraction/complexation was performed in the following way: 10 ppm of 53Cr(III) (concentration of the stock solution: 100 ppm of 53Cr(III)) were complexed with 4 mL EDTA 0.5 mM at pH 10. Standard solutions (53Cr(III) and 50Cr(VI)) together with the chromium-EDTA complex were diluted in HNO3 0.1% (v/v) and injected. It can be observed from Fig. 3a that the separation of the three species (Cr(III)-EDTA−, Cr(III) and Cr(VI)) was achieved. Dead time was 120 s, Cr(III)-EDTA− eluted at first and had a retention time of 178 s, it was followed by Cr(VI) with a retention time of 243 s, and Cr(III) eluted at last with a retention time of 542 s. Contrary to the elution on the AS7 column, on column CS5A, Cr(III)-EDTA− was more retained and appeared 45 s after dead time.

IC-ICP-MS chromatograms obtained through the CS5A column using nitric acid as eluent. a 50 ppb of 53Cr(III), 50 ppb of 50Cr(VI), and 50 ppb of 53Cr(III) in presence of EDTA (after going through the EDTA extraction/complexation conditions), HNO3 0.1% (v/v). b EDTA extraction of a leather sample 5000X dilution in water, spiked with 50 ppb 50Cr(VI). c 53Cr(III) in presence of EDTA (after going through the EDTA complexation conditions). NG1 guard column + CG5A guard column + CS5A column, mobile phase: A: HNO3 1.1 mol/L, B: ultrapure water, flow rate: 0.275 mL/min, KED mode, dead time: 120 s
The order of retention times was the same as the order obtained when using the AS7 ion-exchange column with nitric acid as mobile phase. In fact, the CS5A column has two ion-exchange groups: sulfonic acid (cation-exchange group) and quaternary ammonium (anion-exchange group) [32]. This can explain the order of retention times. Due to the size of the chromium-EDTA complex, it is not strongly retained in the column and it elutes at mild conditions (32% mobile phase A). Cr(VI) anionic species elute when the mobile phase conditions are more acidic (100% mobile phase A) and, therefore, capable of breaking the interactions between the analyte anions and the quaternary ammonium groups of the stationary phase. The Cr(III) cation elutes at the end, 299 s after Cr(VI), as a consequence of the strong interactions with the sulfonic acid groups.
Afterwards, an EDTA extraction of a leather sample was performed. The extract was diluted in water (5000X dilution) and injected. Next, the sample was spiked with 50 ppb of 50Cr(VI) and injected once again. Figure 3 b) corresponds to the spiked sample and it can be noticed that there is a peak at 178 s (the red line denotes the naturally occurring isotope) which corresponds to the chromium-EDTA complex, and a peak at 243 s (the blue line denotes the 50Cr isotope) that corresponds to the spiked 50Cr(VI). The inset from Fig. 3b shows the sample before the 50Cr(VI) spike. It can be concluded from this part that Cr(VI) is indeed present in the EDTA extract but the Cr(III)-EDTA− peak covers the Cr(VI) peak, hence the determination is not possible for samples where there is a large difference in concentrations between Cr(III) (Cr(III)-EDTA− once complexed) and Cr(VI).
In an effort to verify that the Cr(III) complexation with EDTA was complete, 53Cr(III)-EDTA− was diluted and injected separately. As shown in Fig. 3c, when 53Cr(III) is injected in the presence of EDTA after following the extraction protocol, it is possible to observe the main peak of the trivalent chromium complex at 178 s. This permitted to confirm that the Cr(III) complexation with EDTA is complete since no additional peak is observed other than the peak corresponding to Cr(III)-EDTA−.
3.1.4 CS5A column and ammonium nitrate as mobile phase
Separation through the CS5A column was also tested with an ammonium nitrate solution as mobile phase. The chromatographic setup from Fig. 1a was used but a CS5A column replaced the AS7 analytical column, and a CG5A guard column replaced the AG7 guard column. The mobile phase consisted of 75 mmol/L NH4NO3 adjusted to pH 3 at a flow rate of 0.25 mL/min, and the dead time was 175 s. A sample containing 53Cr(III), 50Cr(VI) and 53Cr(III)-EDTA− was injected with an acidic concentration of HNO3 0.1% (v/v). From Fig. 4a, it can be observed that 53Cr(III)-EDTA− elutes just after the dead time with a retention time of 195 s, followed by 50Cr(VI) which elutes at 360 s. No interconversions were appreciated under these conditions but there was no peak for Cr(III) before 3600 s. Even if the CS5A has two ion-exchange groups (sulfonic acid, cation-exchange group, and quaternary ammonium, anion-exchange group) [32], the mobile phase was not strong enough to break the interactions between Cr(III) and the stationary phase. Consequently, Cr(III) remained stuck to the stationary phase and no peak was observed for this cation. Afterwards, a leather sample was extracted through the EDTA extraction method. The concentrated extract was posteriorly injected and a peak was observed at 195 s which corresponds to Cr(III)-EDTA−. Additionally, a small peak was observed at 360 s which corresponds to Cr(VI). Nevertheless, coelution of both peaks impede the quantification of both species, as it can be appreciated from the inset on Fig. 4b.

IC-ICP-MS chromatograms obtained through the CS5A column using ammonium nitrate as eluent. a 100 ppb of 53Cr(III), 100 ppb of 50Cr(VI) and 100 ppb of 53Cr(III) in presence of EDTA (after going through the EDTA extraction/complexation conditions), HNO3 0.1% (v/v). b EDTA extraction of a leather sample, no dilution. NG1 guard column + CG5A guard column + CS5A column, mobile phase: 75 mmol/L NH4NO3 pH 3, flow rate: 0.25 mL/min, STD mode, dead time: 175 s
3.1.5 AS7 column and ammonium nitrate as mobile phase (optimal conditions)
In the literature, there are limited studies related to chromium speciation in leathers.
Concerning other matrices, Gürleyük and Wallschläger [33] derivatized trivalent chromium with EDTA and achieved its separation from hexavalent chromium in certified reference materials (CRMs) (synthetic and estuarine water) by IC-ICP-MS using a self-regenerating suppressor to avoid salt deposition, an anion-exchange AS7 column (4 × 250 mm, particle size: 10 µm), an AG7 guard column (4 × 50 mm, particle size: 10 µm), and 50 mM NH4NO3 at pH 8 as chromatographic eluent. They obtained an Limit of detection (LOD) of 12 ng/L for Cr(VI) and 5 ng/L for Cr(III).
Encouraged by the reported results, the chromatographic setup from Fig. 1a was used (NG1 and AG7 guard columns, and an AS7 analytical column). Additionally, further research was done and pH of the mobile phase was adjusted to 3 to prevent reduction of Cr(VI) as well as the precipitation of Cr(III). According to the diagram of species distribution (Additional file 1: Figure S2), there is only a small pH window (shaded area) where Cr(VI) and Cr(III) species are present in ionic form. Above pH 4, Cr(III) forms insoluble hydroxo-aqua complexes; pH values below 2 may result in the formation of H2CrO4, which is a strong oxidizing agent [34]. The concentration of the mobile phase was also modified (75 mM NH4NO3 at pH 3, adjusted with HNO3 1 M) and interconversions were followed with the aid of chromium isotopic standards.
The first results obtained under these conditions and working with chromium isotopic standards where published on a previous study [17]. The main observations where the following:
-
When a 50Cr(VI) standard solution was injected only one peak was seen at 920 s, which corresponds to the elution of Cr(VI).
-
When a 53Cr(III) standard solution was injected in the presence of EDTA after having gone through the EDTA extraction conditions, the peak of the trivalent chromium complex appeared at 230 s. No other peak was observed.
-
When a 50Cr(VI) standard solution was injected in the presence of EDTA, it did not complex with EDTA, it remained as 50Cr(VI) but a small part was transformed into 50Cr(III), which complexed with EDTA (50Cr(III)-EDTA−), and this represented only 1.5% of the main peak (the 50Cr(VI) peak). This is not a major problem because trivalent chromium is present in leather in much larger quantities than hexavalent chromium, so a minimal conversion of Cr(VI) to Cr(III) represents a negligible amount of the total amount of Cr(III).
-
No peak was observed when injecting a 53Cr(III) standard solution, probably because it was too strongly linked to the sulfonic group of the core of the column.
Later on, different samples were injected in order to evaluate the retention times of chromium peaks observed at different pH and redox potentials (dead time = 170 s). Table 5 summarizes the obtained results. It is important to highlight the following remarks:
-
1.
At a sample pH of 3.00, complexation does not take place. However, complexation of the standards was achieved at pH 9.57. This can be explained by the fact that at low pH (acidic conditions), the protonated form of EDTA is prevalent and H+ competes with the metal ion, Cr3+, for EDTA. At basic conditions, the deprotonated form of EDTA is prevalent and complexation between Cr3+ and EDTA occurs rapidly.
-
2.
After 50Cr(VI) and 53Cr(III) went through the EDTA extraction conditions at pH 4.14, 50Cr(VI) is transformed into 50Cr(III) which complexed with EDTA. This is possible because heating accelerates the kinetics of chromium’s complexation with EDTA. In fact, once complexed, Cr(III)-EDTA− is stable in a wide range of pH but complexation must be performed at pH 10 in order to preserve the oxidation state of Cr(VI), because heating at acidic pH promotes its reduction to Cr(III).
-
3.
When the undiluted leather extract was spiked with 50Cr(VI) and then injected, the 50Cr(VI) peak appeared at 420 s. However, when the diluted leather extract was spiked with 50Cr(VI) and subsequently injected, the 50Cr(VI) peak appeared at 970 s. Thus, when Cr(VI) is present in an undiluted EDTA leather extract, it is probably bonded to something that can be found in the leather matrix. On the other hand, when Cr(VI) is present in a diluted sample, the conditions are closer to when Cr(VI) is alone in an aqueous solution, therefore the retention time remains the same. Another hypothesis is that Cr(VI) is present in different ionic forms when the sample is diluted or not diluted, hence, Cr(VI) elutes at two different retention times.
-
4.
It is important to mention that an EDTA solution was also injected after the injection of real samples and standards, and no peaks were observed. This permitted to confirm that there was as total elution of the compounds.
After all the experiments performed, it was possible to confirm that complexation should take place at high pH (around 10) and then Cr(III)-EDTA− is stable under a large range of pH with no shifting of the peak depending on the dilution factor. Therefore, the EDTA extraction with further analysis by IC-ICP-MS was feasible for trivalent chromium determination.
By the other hand, the Cr(VI) peak may shift depending on the dilution factor of the leather extract (EDTA extraction). Consequently, the extraction of Cr(VI) from leather samples through the ISO 17075 [13] protocol with subsequent analysis through IC-ICP-MS was tested. Chromium isotopic standards as well as a leather sample followed the whole ISO 17075 protocol with the aim of testing for possible interconversions.
As it can be seen from Fig. 5a, when injecting 53Cr(III) after following the whole ISO 17075 protocol, it does not transform into 53Cr(VI). Moreover, when injecting 50Cr(VI), which had followed the whole ISO 17075 protocol, a single peak appeared at 925 s (Fig. 5b). When injecting an ISO leather extract, a small peak was observed at 925 s, which corresponds to the retention time of Cr(VI) (Fig. 5c). Therefore, the ISO standard method can be used to extract Cr(VI) from leather samples and the extract can be then analyzed through IC-ICP-MS under the optimized analysis conditions and no interconversions occur.

Chromatograms obtained after ISO 17075 extraction. a 50 ppb of 53Cr(III) after going through the ISO 17075 extraction. b 50 ppb of 50Cr(VI) after going through the ISO 17075 extraction. c Undiluted leather extract (through ISO 17075). NG1 guard column + AG7 guard column + AS7 column, mobile phase: 75 mmol/L NH4NO3 pH 3, flow rate: 0.25 mL/min, KED mode, dead time: 170 s
3.2 FTICR MS
As mentioned before, the ESI source was used in positive and negative ion modes, but the negative ionization mode provided the highest intensity for Cr(III)-EDTA−, as compared to the positive ion mode. Therefore, only the spectra for the negative ion mode are presented.
The negative ion mode ESI FTICR mass spectrum of 10 ppm of Cr(III) in presence of EDTA 0.5 mmol/L (after going through the EDTA extraction/complexation conditions) (2X dilution in ACN) is presented in Additional file 1: Figure S3 a). This spectrum shows the deprotonated EDTA ion, [EDTA − H]−, at m/z 291.08339, as well as the deprotonated metal-EDTA complex ion, [EDTA − 4H + Cr(III)]−, at m/z 340.00042, and the peak that corresponds to the loss of formic acid from the EDTA molecule, [EDTA–H–HCOOH]−, at m/z 245.07807.
Likewise, the negative ion mode ESI FTICR mass spectrum of an EDTA leather extract (2X dilution in ACN/H2O, 50/50 v/v%) which is presented in Additional file 1: Figure S3b shows the deprotonated EDTA ion, [EDTA \(-\) H]−, present at m/z 291.08339, as well as the deprotonated metal-EDTA complex ion, [EDTA − 4H + Cr(III)]−, at m/z 340.00042, and the peak that corresponds to the loss of formic acid from the EDTA molecule, [EDTA–H–HCOOH]−, at m/z 245.07782.
It is important to mention that no peaks were found that suggest the formation of clusters of more than one metal ion per EDTA molecule, or vice versa, the formation of clusters of more than one EDTA molecule per metal ion.
A spectrum of 10 ppm of Cr(III) in presence of EDTA 0.5 mmol/L (after going through the EDTA extraction/complexation conditions) (2X dilution in ACN), as well as a spectrum of the isotope pattern simulation of Cr(III)-EDTA− can be found in Additional file 1: Figure S4. From this figure it can be observed that the simulation pattern matches the spectrum obtained experimentally. Due to the difference in natural abundances of chromium naturally occurring isotopes (50Cr: 4.345%, 52Cr: 83.789%, 53Cr: 9.501%, 54Cr: 2.365%), the following peaks are observed (m/z are rounded to the nearest unit. For exact values, see Additional file 1: Figure S4):
-
m/z 338: [EDTA–4H + 50Cr(III)]−
-
m/z 340: [EDTA–4H + 52Cr(III)]−
-
m/z 341: [EDTA–4H + 53Cr(III)]−
-
m/z 342: [EDTA–4H + 54Cr(III)]−
Table 6 shows the ion formula prediction for the peaks observed at m/z 291 and 340, at negative ion mode, and the peaks observed at m/z 293 and 342, at positive ion mode. The low error value permitted to confirm the formula of the chromium-EDTA complex: C10H13CrN2O8 (Cr(III)-EDTA−).
3.3 Validation
Once the extraction and analytical conditions were optimized, the method was validated for Cr(III) and Cr(VI). For a given sample, Cr(III) was extracted via the EDTA extraction as previously specified, and Cr(VI) was extracted via the ISO 17075 protocol. Both extracts were analyzed with the same setup under the same analytical conditions (Sect. 3.1.5). This ensured an accurate and simultaneous determination of both species, even at a large concentration difference between Cr(III) and Cr(VI), as is the case for the actual leather samples under study, and without interconversions.
As it has been proved by previous studies [17], EDTA is the most performant method for the quantitative extraction of Cr(III) as a chromium-EDTA complex (Cr(III)-EDTA−). Furthermore, under the optimized analysis conditions (75 mmol/L NH4NO3 at pH 3), Cr(III)-EDTA− is stable under a large range of sample pH with no shifting of the peak depending on the dilution factor.
This method was validated in terms of linearity, LOD, LOQ and interday precision. Linearity was studied through the analysis of calibration solutions of Cr(III)-EDTA− from 0 to 100 \(\upmu\)g/L of Cr(III) (n = 6, 3 replicate injections per point). In order to do this, 400 \(\upmu\)L of a Cr(III) solution were added to 4 mL of an EDTA solution (0.5 mM at pH 10). Once the microwave assisted complexation was completed, the solution containing the complex was diluted in water and each standard was analyzed through IC-ICP-MS, focusing on the peak with a retention time of 250 s, which corresponds to the chromium-EDTA complex. Peak area was plotted against concentration, and R2 was found to be 0.9990. LOD and LOQ were calculated as the concentration necessary to give a signal-to-noise (S/N) ratio equal to 3 and 10, respectively. LOD (S/N = 3) was found to be 0.016 \(\upmu\)g/L or 0.3 \(\upmu\)g/Kg for Cr(III), and LOQ (S/N = 10) was found to be 0.054 \(\upmu\)g/L or 1.1 \(\upmu\)g/Kg for Cr(III).
Interday precision was determined by analyzing three different leather samples (Sample 1: natural E2, Sample 2: after polish, Sample 3: UV-aging) from which trivalent chromium was extracted on three different days. As it can be seen in Table 7, for each sample the interday precision provided a relative standard deviation (RSD) below 12% and a low standard deviation (SD). Low RSD values show that the method exhibits excellent interday precision.
As mentioned before, the ISO 17075 [13] standard method was used to extract Cr(VI) from leather samples and the extract was then analyzed through IC-ICP-MS under the optimized analysis conditions and no interconversions were observed.
This method was validated in terms of linearity, LOD, LOQ and precision. Linearity was studied through the analysis of calibration solutions of Cr(VI) from 0 to 100 \(\upmu\)g/L (n = 6, 3 replicate injections per point). Each standard was analyzed through IC-ICP-MS, focusing on the peak with a retention time of 960 s, which corresponds to Cr(VI). Peak area was plotted against concentration, and R2 was found to be 0.9993.
LOD and LOQ were calculated as the concentration necessary to give a S/N ratio equal to 3 and 10, respectively. LOD (S/N = 3) was found to be 0.13 \(\upmu\)g/L or 7 \(\upmu\)g/Kg for Cr(VI), and LOQ (S/N = 10) was found to be 0.43 \(\upmu\)g/L or 22 \(\upmu\)g/Kg for Cr(VI).
Precision was determined by analyzing three different leather samples (Sample 1: UV-aging, Sample 2: mirage, Sample 3: after polish) from which hexavalent chromium was extracted three times the same day through the ISO 17075 [13] protocol. As it can be seen in Table 8, RSD was below 5.3%. Low RSD values show that the method exhibits excellent precision.
4 Conclusions
This work focused on the analysis of trivalent and hexavalent chromium extracted from leather samples. The challenge was to extract and quantify both chromium species, with high concentration difference between Cr(III) and Cr(VI), without interconversions during the extraction and/or the analysis conditions. Separation and speciation were studied by IC-ICP-MS. Several conditions were tested such as: two different columns, different mobile phases and different pH of the samples. Among all these tests, the optimized conditions for the analysis of chromium species were: a NG1 guard column, an AG7 guard column, an AS7 ion-exchange column, and 75 mmol/L NH4NO3 pH 3 as chromatographic eluent at a flow rate of 0.25 mL/min. LOD and LOQ for Cr(III) were found to be 0.016 and 0.054 \(\upmu\)g/L (0.3 and 1.1 \(\upmu\)g/Kg), respectively, and for Cr(VI) 0.13 and 0.43 \(\upmu\)g/L (7 and 22 \(\upmu\)g/Kg), respectively. Even if the other tested conditions were not suitable for leather samples, they served to prove that the EDTA extraction of Cr(III) was complete, as long as the extraction is performed at basic conditions. FTICR-MS allowed the structure elucidation of the chromium-EDTA complex formed during the EDTA extraction. Interconversions were monitored with the use of chromium isotopic standards. Regarding the extraction methods, EDTA proved to be the most performant method for the extraction of Cr(III), and ensured Cr(VI) extraction but a small amount (1.5%) of Cr(VI) could be converted into Cr(III). Consequently, ISO 17075 protocol was used for the extraction of Cr(VI). In such case, both extracts (the sample extracted through the EDTA extraction and the same sample extracted through the ISO 17075 protocol) could be analyzed with the same setup under the same analytical conditions without any interconversion.
Availability of data and materials
The datasets analyzed during the current study are available from the corresponding author on reasonable request.
Change history
15 June 2023
A few of the units μg were mistakenly typed as gμ, which have been updated correctly.
Abbreviations
- ACN:
-
Acetonitrile
- CRMs:
-
Certified reference materials
- DPC:
-
1,5-Diphenylcarbazide
- DTPA:
-
Diethylenetriaminepentaacetic acid
- EDTA:
-
Ethylenediaminetetraacetic acid
- FTICR:
-
Fourier-transform ion cyclotron resonance mass spectrometry
- HPLC-ICP-MS:
-
High-performance liquid chromatography-inductively coupled plasma-mass spectrometry
- IC-ICP-MS:
-
Ion chromatography-inductively coupled plasma-mass spectrometry
- ICP-MS:
-
Inductively coupled plasma-mass spectrometry
- ISO:
-
International Organization for Standardization
- KED:
-
Kinetic energy discrimination mode
- LOD:
-
Limit of detection
- LOQ:
-
Limit of quantification
- R2 :
-
Coefficient of determination
- RSD:
-
Relative standard deviation
- S/N:
-
Signa-to-noise
- SD:
-
Standard deviation
- STD:
-
Standard mode
References
Dias da Silva LI, Marinho Pontes FV, Castro Carneiro M, Couto Monteiro MI, Dominguez de Almeida M, Alcover Neto A. Evaluation of the chromium bioavailability in tanned leather shavings using the SM&T sequential extractions scheme. Chem Speciat Bioavailab. 2011;23:183–7.
Ertani A, Mietto A, Borin M, Nardi S. Chromium in agricultural soils and crops: a review. Water Air Soil Pollut. 2017;228:1–12.
Covington AD, Wise WR. Tanning chemistry: the science of leather. 2nd ed. London: Royal Society of Chemistry; 2019.
Dixit S, Yadav A, Dwivedi PD, Das M. Toxic hazards of leather industry and technologies to combat threat: a review. J Clean Prod. 2015;87:39–49.
Arellano-Sánchez MG, Devouge-Boyer C, Hubert-Roux M, Afonso C, Mignot M. Chromium determination in leather and other matrices: a review. Crit Rev Anal Chem. 2021;52:1–20.
Fontaine M, Clement Y, Blanc N, Demesmay C. Hexavalent chromium release from leather over time natural ageing vs accelerated ageing according to a multivariate approach. J Hazard Mater. 2019;368:811–8.
Fontaine M. Étude de l’évolution de la teneur en chrome hexavalent dans le cuir, influence des conditions de fabrication et importance des facteurs environnementaux [Internet]. [Lyon]: Université de Lyon; 2017. https://tel.archives-ouvertes.fr/tel-01684756.
Hedberg YS, Erfani B, Matura M, Lidén C. Chromium(III) release from chromium-tanned leather elicits allergic contact dermatitis: a use test study. Contact Derm. 2018;78:307–14.
Hedberg YS. Chromium and leather: a review on the chemistry of relevance for allergic contact dermatitis to chromium. J Leather Sci Eng. 2020;2:15.
Commission regulation (EU) No 301/2014 amending annex XVII to regulation (EC) No 1907/2006 of the European parliament and of the council on the registration, evaluation, authorisation and restriction of chemicals (REACH) as regards chromium VI compounds. Official Journal of the European Union. 2014 Mar 25;3.
OEKO-TEX® - International association for research and testing in the field of textile and leather ecology. Leather standard by OEKO‑TEX® [Internet]. OEKO-TEX® Service GmbH; 2021 [cited 2022 Jun 20]. https://www.oeko-tex.com/importedmedia/downloadfiles/LEATHER_STANDARD_by_OEKO-TEX_R__-_Standard_en.pdf.
International organization for standardization. ISO 17075-1:2017 [IULTCS/IUC 18–1] Leather—Chemical determination of chromium(VI) content in leather—Part 1: Colorimetric method [Internet]. 2017. https://www.iso.org/standard/67096.html.
International organization for standardization. ISO 17075-2:2017 [IULTCS/IUC 18-2] Leather—Chemical determination of chromium(VI) content in leather—Part 2: Chromatographic method [Internet]. 2017. https://www.iso.org/standard/67097.html.
Pastore P, Favaro G, Ballardin A, Danieletto D. Evidence of Cr(VI) formation during analysis of leather. Talanta. 2004;63:941–7.
Rezić I, Zeiner M. Determination of extractable chromium from leather. Monatsh Chem. 2009;140:325–8.
Costa V, Neiva A, Pereira-Filho E. Chromium speciation in leather samples: an experiment using digital images, mobile phones and environmental concepts. Ecletica Quim J. 2019;44:62–74.
Arellano-Sánchez MG, Devouge-Boyer C, Hubert-Roux M, Afonso C, Mignot M. Quantitative extraction of chromium VI and III from tanned leather: a comparative study of pretreatment methods. J Leather Sci Eng. 2021;3:30.
Hernandez F, Séby F, Millour S, Noël L, Guérin T. Optimisation of selective alkaline extraction for Cr(VI) determination in dairy and cereal products by HPIC–ICPMS using an experimental design. Food Chem. 2017;214:339–46.
Hernandez F, Jitaru P, Cormant F, Noël L, Guérin T. Development and application of a method for Cr(III) determination in dairy products by HPLC–ICP-MS. Food Chem. 2018;240:183–8.
Hamilton EM, Young SD, Bailey EH, Watts MJ. Chromium speciation in foodstuffs: a review. Food Chem. 2018;250:105–12.
Petrucci F, Senofonte O. Determination of Cr(VI) in cosmetic products using ion chromatography with dynamic reaction cell-inductively coupled plasma-mass spectrometry (DRC-ICP-MS). Anal Methods. 2015;7:5269–74.
Hu L, Cai Y, Jiang G. Occurrence and speciation of polymeric chromium(III), monomeric chromium(III) and chromium(VI) in environmental samples. Chemosphere. 2016;156:14–20.
Mädler S, Todd A, Skip Kingston HM, Pamuku M, Sun F, Tat C, et al. Ultra-trace level speciated isotope dilution measurement of Cr(VI) using ion chromatography tandem mass spectrometry in environmental waters. Talanta. 2016;156–157:104–11.
Jabłońska-Czapla M. Arsenic, antimony, chromium, and thallium speciation in water and sediment samples with the LC-ICP-MS technique. Int J Anal Chem. 2015;2015:1–13.
Li P, Li L, Xia J, Cao S, Hu X, Lian H-Z, et al. Determination of hexavalent chromium in traditional Chinese medicines by high-performance liquid chromatography with inductively coupled plasma mass spectrometry: liquid chromatography. J Sep Sci. 2015;38:4043–7.
Martone N, Rahman GMM, Pamuku M, Skip Kingston HM. Determination of chromium species in dietary supplements using speciated isotope dilution mass spectrometry with mass balance. J Agric Food Chem. 2013;61:9966–76.
Kutscher D, McSheehy S, Wills J, Jensen D. Speciation analysis of Cr (III) and Cr (VI) in drinking waters using anion exchange chromatography coupled to the thermo scientific iCAP Q ICP-MS [Internet]. Thermo Fisher Scientific; 2016. https://assets.thermofisher.com/TFS-Assets/CMD/Application-Notes/AN-43098-ICP-MS-Chromium-Drinking-Water-AN43098-EN.pdf.
Rabin S, Stillian J. Practical aspects on the use of organic solvents in ion chromatography. J Chromatogr A. 1994;671:63–71.
DionexTM IonPacTM AS7 IC columns [Internet]. [cited 2022 Jun 12]. https://www.thermofisher.com/order/catalog/product/035393.
Guillarme D, Nguyen DTT, Rudaz S, Veuthey JL. Method transfer for fast liquid chromatography in pharmaceutical analysis: application to short columns packed with small particle. Part I: isocratic separation. Eur J Pharm Biopharm. 2007;66:475–82.
Guillarme D, Nguyen DT-T, Rudaz S, Veuthey J-L. Method transfer for fast liquid chromatography in pharmaceutical analysis: application to short columns packed with small particle. Part II: Gradient experiments. Eur J Pharm Biopharm. 2008;68:430–40.
DionexTM IonPacTM CS5A IC columns [Internet]. [cited 2022 Jun 12]. https://www.thermofisher.com/order/catalog/product/046100TS.
Gürleyük H, Wallschläger D. Determination of chromium(III) and chromium(VI) using suppressed ion chromatography inductively coupled plasma mass spectrometry. J Anal At Spectrom. 2001;16:926–30.
Hagendorfer H, Goessler W. Separation of chromium(III) and chromium(VI) by ion chromatography and an inductively coupled plasma mass spectrometer as element-selective detector. Talanta. 2008;76:656–61.
Acknowledgements
Not applicable.
Funding
This work has been partially supported by University of Rouen Normandy, INSA Rouen Normandy, the Centre National de la Recherche Scientifique (CNRS), European Regional Development Fund (ERDF HN0001343), Labex SynOrg (ANR-11-LABX-0029), Carnot Institute I2C, the graduate school for research XL-Chem (ANR-18-EURE-0020 XL CHEM), Region Normandie, and Consejo Nacional de Ciencia y Tecnología (CONACYT CVU 707668).
Author information
Authors and Affiliations
Contributions
MGAS performed the extractions, analyzed, and interpreted the data. JV and LM participated in the analysis of the extracts. CDB and MHR were in charge of the instrumentation. CA was in charge of getting the funding and provision of equipment. MM corrected the manuscript and leaded scientific discussions. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Figure S1. Summary of the conditions tested for the speciation analyses. Figure S2. Distribution of chromium species in water. Figure S3. Negative ion mode ESI FTICR mass spectra. a) 10 ppm of Crin presence of EDTA 0.5 mmol/L. b) EDTA leather extract. Figure S4. Negative ion mode ESI FTICR mass spectra. a) 10 ppm of Crin presence of EDTA 0.5 mmol/L. b) Isotope pattern simulation of Cr-EDTA−.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Arellano-Sánchez, M.G., Vievard, J., Moufarrej, L. et al. Separation, speciation and quantification of both chromium (VI) and chromium (III) in tanned leather samples: a comparative study and validation of analytical methods. Collagen & Leather 5, 18 (2023). https://doi.org/10.1186/s42825-023-00125-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42825-023-00125-7