
Abstract
Background
Brucellosis is a bacterial zoonosis of public health and economic importance worldwide. It affects a number of domestic animals, wild animals and humans. Human brucellosis originates from either livestock or wildlife. The species of Brucella circulating in wild animals in Tanzania is largely unknown due to insufficient surveillance. This study was carried out to identify Brucella species found in selected wildlife hosts in the Serengeti ecosystem.
Methodology
The study used a total of 189 archived samples that were obtained from cross-sectional studies previously conducted between 2000 and 2017 in the Serengeti ecosystem in Tanzania. Whole blood, serum and amniotic fluid collected from buffalos, lions, wildebeest, impala, zebra and hyena were available for DNA extraction. Multiplex polymerase chain reaction for B. abortus, B. melitensis, B. ovis and B. suis (AMOS PCR) and quantitative real-time PCR (qPCR) targeting the bcsp31 and IS711 genes for Brucella genus detection and the IS711 targets alkB for B. abortus and BMEI1162 for B. melitensis were used to detect Brucella strains.
Results
Out of the 189 samples tested, 12 (6.35 %) and 22 (11.6 %) were positive to AMOS-PCR and qPCR, respectively. Most of the positive samples were from lions (52.6 %) and buffaloes (19.6 %). Other animals that were positive included: wildebeest (13.6 %), impala (13.6 %), zebra (4.5 %) and hyena (4.5 %). Out of 22 positive samples, 16 (66.7 %) were identified as B. abortus and the other six samples did not amplify for neither B. abortus nor B. melitensis.
Conclusions
The detection of Brucella DNA in archived wild animal samples shows testing potential of samples collected from this population. The zoonotic species B. abortus and B. melitensis detected in wild animals have previously been reported in livestock and humans in the region. The findings suggest that, due to the contact network, some of the identified wild animal hosts in this study could be reservoirs for infections in domestic animals and humans within the Serengeti ecosystem while others are likely dead-end hosts. One Health control strategies and continuous surveillance programs in other wildlife reserved areas should be implemented to help predicting transmission in livestock and humans in the region.
Similar content being viewed by others
Introduction
Brucellosis affects a number of domestic and wild animal species as well as humans [1, 2]. The disease is a public health problem that is challenging to control in many developing countries including Tanzania, especially in pastoral and agro-pastoral farming systems [3,4,5]. According to the World Health Organization (WHO), brucellosis is an important re-emerging, neglected tropical zoonosis [6] largely due to lack of awareness, and minimal investment in surveillance and control measures.
In wild animals, brucellosis occurs as a result of spill-over from infected livestock or as a natural, sustained infection within susceptible wild animal populations [7, 8]. Wild ungulates can acquire infection by ingesting contaminated forage [8]. Carnivores such as wolves (Canis lupus) and foxes (Vulpes vulpes) are thought to be exposed through the ingestion of infected animals, placentae or aborted fetuses [2]. The disease has been reported in wild animals in some African countries, including Kenya [9], South Africa [8], Zimbabwe [10] and Tanzania [11,12,13,14]. Among Tanzanian wild animals, Brucella infections have been reported in topi (Damaliscus lunatus jimela), buffalo (Syncerus caffer), impala (Aepyceros melampus), Thompson gazelle (Eudorcas thomsonii) and wildebeest (Connochaetes) [15, 16]. However, most of these reports were based on serological studies, without identification of the Brucella spp. involved. Other studies reported brucellosis in livestock-wildlife interfaces in the Ngorongoro Conservation Area [17] and the Mikumi-Selous Ecosystem [18].
In recent years, many African countries have prioritized zoonotic diseases under the Global Health Security Agenda and brucellosis has been ranked among priority zoonotic diseases for control [19, 20]. In Tanzania, in particular, brucellosis ranks sixth among the priority zoonoses that the country focuses its control efforts on [16, 21]. Since the prioritization of brucellosis in 2017, a number of efforts for control, including development of a national control strategy, enacted vaccine and vaccination regulation and vaccination campaigns have been put in place. Critically highlighted areas include the contribution of different hosts to the transmission and maintenance of the disease in the country [16]. Studies on brucellosis in Tanzania have shed light on Brucella species circulating in the different livestock species within different regions [17, 18, 22]. However, Brucella spp. strains circulating in wild animal populations remain under-reported [16, 21]. The aim of this study was to identify the Brucella species circulating among wild animals in the Serengeti ecosystem in Tanzania, using molecular techniques and to evaluate the usefulness of archived samples in yielding information on circulating Brucella spp. By using clinical/field samples archived for up to 15 years, the study sought to detect and characterize Brucella DNA extracted directly from samples, most of which were not viable for bacteriological culture and/ or serological exploration.
Materials and methods
Study area
The samples used were collected during various cross-sectional studies previously conducted in the Serengeti ecosystem in Tanzania. The Serengeti ecosystem is located in the northwest of the country between the Ngorongoro highlands and Lake Victoria. This ecosystem comprises of Serengeti National Park, the Ngorongoro Conservation Area, Maswa Game Reserve, Loliondo Game Controlled Area and Kenya’s Masai Mara National Reserve (Fig. 1). The study area was selected because there is notable interaction between wild animals, livestock, and humans. The area is mainly inhabited by the Maasai, with livestock keeping as their main socio-economic activity [23]. Furthermore, there have been previous reports on brucellosis in humans and livestock around the ecosystem [22, 24, 25].

A map of the study area showing the Serengeti ecosystem [26]
Collection of biological samples
All samples were retrieved from the archive of the Tanzania Wildlife Research Institute (TAWIRI) bio-repository in Arusha and the Serengeti laboratory. Whole blood (collected in the EDTA tubes), serum and amniotic fluid samples were used in the present study. The samples were collected between 2000 and 2017 during routine surveillance, research and veterinary training programs. From TAWIRI and the Serengeti laboratory, all samples were transported in cold chain and stored at -20 °C at the microbiology laboratory, college of Veterinary Medicine and Biomedical Sciences in Sokoine University of Agriculture (SUA) Tanzania. The retrieved samples were originally collected from buffaloes, wildebeest, zebra, lions, baboon, impala and hyena. These were the only samples available for this study. In total, 189 samples were used, out of which 11 were amniotic fluid, 170 whole blood and eight serum samples.
Molecular detection of Brucella spp
The study employed a conventional AMOS PCR for the detection of B. abortus biovars 1, 2, and 4, B. melitensis biovars 1, 2, and 3, B. ovis and B. suis biovars 1, 2, 3, 4 and 5. A quantitative real -time PCR (qPCR) for the detection of Brucella spp. from DNA extracts was also used. DNA extraction from samples was done at the microbiology laboratory, college of Veterinary Medicine and Biomedical Sciences in SUA Tanzania. A commercial DNA extraction kit (Zymo Research, USA Genomic DNA Tissue Mini Prep kit) was used as previously described [27]. Briefly, 40 µl of genomic lysis buffer were added to 200 µl of the source sample. The mixture was subjected to digestion, deactivation, washing and elution steps as per manufacturer’s instructions. Stock DNA samples were stored at -20 ºC until PCR was performed.
The conventional AMOS PCR was run as previously described [28]. Briefly, a reaction mixture of a final concentration of 0.5µM for each of the primers (forward and reverse), 5 µl of the DNA template and x1 concentration of the OneTaq Quick-Load DNA Polymerase PCR master mix (New England BioLabs, Mass., USA) were prepared up to a final volume of 25 µl. After an initial denaturization step of 5 min at 95 °C in a thermo cycler (TaKaRa, Japan), the mixture underwent 35 cycles of denaturization at 94 °C for 1 min. Annealing at 53 °C for 30 s, extension at 72 °C for 7 min, and final extension steps at 72 °C for 10 min were then performed. Amplification of the target region was confirmed based on the presence of specific bands for the different Brucella spp. The PCR products (3 µl) were analyzed on a 1.5 % agarose gel pre-stained with bromide dye (Invitrogen Carlsbad, CA) run at 100 V for 60 min for electrophoresis detection and direct visualization. The primers used in this analysis were obtained from Bioline Inc (Taunton, MA, USA) as previously described [28].
The qPCR for Brucella genus identification targeted bcsp31 and IS711 gene regions as previously described [29, 30]. All qPCR assays were run on the Premier instruments (Biosoft International, Palo, Alto, Calif.) at a final volume of 25 µl, consisting; 12.5 µl of 2 X master mix, 2.5 µl of purified DNA template, 2.5 µl of internal positive control (IPC) master mix and 0.5 µl of IPC synthetic DNA from the Luna Universal Probe One-Step real-time qPCR premix kit (New England BioLabs, Mass., USA). After an initiation at 50ºC for 2 min and denaturation 95ºC for 10 min, activation of the polymerase enzyme followed by 40 cycles of: 95 ºC for 15 s, and 60 ºC for 1 min thermocycling, repeated for approximately 100 min. Samples were considered positive only if they amplified in both bcsp31 and IS711 targets and below a predetermined cycle time (< 39).
Samples positive for the Brucella genus level target were then subjected to a multiplex assay to distinguish B. abortus from B. melitensis. The assay used B. abortus and B. melitensis primers targeting the insertion sequences downstream of alkB and BMEI1162 targets respectively [30]. Analysis was done according to manufacturer instruction in the Brucella genus Genesig standard kit (Genesig Camberly, UK). A volume of 10 µl DNA was mixed with primers and probes in 1000 µl reaction tubes as detailed in Probert et al. [30]. Primers and probes used in the qPCR assay for the detection of Brucella spp. are described elsewhere [29]. Amplification and real-time fluorescence detection were performed on the iCycler real-time PCR detection system (Bio-Rad Laboratories, Hercules, Calif.).
The results from each of the techniques were collated in Microsoft Excel then descriptive and analytical statistics were done using R software [31]. A positive result was considered only if a sample was positive by both qPCR assays, or by the AMOS PCR. Proportions of positivity by the qPCR assay were then estimated for each category of variables.
Cross tabulation was used to determine the diagnostic sensitivity and specificity of the AMOS and real-time qPCR using the qPCR speciation assay as the reference test.
Results
Samples from seven wild animal species, buffaloes, wildebeest, zebra, lions, baboons, impala and hyenas, were used in the present study. Out of 189 identified samples, 170 were whole blood collected in EDTA tubes, eight were sera and 11 amniotic fluid samples. Most of the samples (80; 42.3 %) were from wildebeest, and a larger proportion of the samples (183; 96.8 %) were also obtained from female animals. In terms of specific location, the majority (115; 60.9 %) of samples were from the Serengeti National Park while the rest were from the Ngorongoro conservation area and National Park. It was found that the age of the wild animal sampled (adult), location (Serengeti) and the sample type used for DNA extraction (whole blood) were all significantly associated with the detection of Brucella DNA (Table 1).
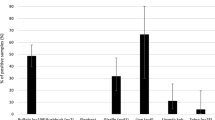
Of the 189 samples screened, Brucella DNA was identified in 12 (6.3 %) samples (nine whole blood, one serum and two amniotic fluid) based on AMOS PCR (Supplementary material S1). Out of the 12 positive samples, Brucella species identified included four B. abortus, one B. melitensis and six B. suis and one was unidentified (Fig. 2). The animal species distributions of Brucella DNA positive samples based on AMOS PCR and the qPCR speciation assay are detailed in Table 2.

Schematic aggregate plot for the samples tested positive for the three assays (AMOS PCR, qPCR genus specific and species specific), Brucella spp. identified and the respective agreement across tests
The qPCR test results indicated that 22 samples (11.6 %) were positive for Brucella DNA. Overall, 16 samples out of 22 (72.7 %) samples were positive for B. abortus in the real-time qPCR speciation assay and six samples did not amplify for either species (Fig. 2). The 16 samples that were positive for B. abortus included two samples that were also positive for the same species in AMOS PCR. Using the real-time speciation assay as the reference test, AMOS PCR had a sensitivity of 16.7 % and specificity of 92 %, while the qPCR assay had a sensitivity of 72.7 % and specificity of 100 % (Table 3). The full data set for samples positive by all the tests are shown in the supplementary material (S2).
Discussion
The purpose of the current study was to determine the Brucella species circulating among wild animals in the Serengeti ecosystem in Tanzania, using molecular techniques and to evaluate the usefulness of archived samples yielding information on Brucella spp. Zoonotic Brucella spp. were detected in wild animal populations in the Serengeti ecosystem using qPCR and AMOS PCR. Lions and buffaloes had the highest proportions of positivity from the sample pool. The most identified species in the wild animals was Brucella abortus although B. melitensis was also detected. This is the first reported study to conduct molecular detection of Brucella directly from archived samples of wild animals from Africa. Detecting Brucella circulating in the blood is quite rare and that this detection method underestimates infection rates because Brucella is hiding out in other tissues since this is an intracellular parasite. However, other studies have indicated serum as a preferred sample source for Brucella detection [32].
Results obtained from qPCR show that B. abortus was dominant in the samples collected suggesting that it is a common Brucella species circulating in the Serengeti ecosystem. Detection of Brucella spp. from the study area is not surprising, as previous studies have reported Brucella sero-positivity that ranged between 10.5 and 17 % [14] in wild animals in Tanzania including the Serengeti ecosystem [12, 22]. Therefore, detection of pathogenic DNA in samples collected from wild animals in the study area, further confirms that Brucella is circulating in the ecosystem.
It was further observed that Brucella DNA were detected more in lions (25 % by AMOS PCR and 38 % by qPCR) or buffaloes (8.3 % by AMOS PCR and 43.8 % by qPCR) than in other wild animal species. It is probably, that this was the case because lions are indiscriminate carnivores and are likely to prey on Brucella infected animals like buffaloes [15, 33]. However, the high detection rates observed in buffaloes may be due to B. abortus being the common species in the ecosystem and is known to mostly infect bovine ungulates [33]. Generally, detection of zoonotic Brucella in wild animals in this study, especially B. melitensis and B. abortus that have already been found in the region, points to the possibility that they are the source of sustained Brucella transmission in livestock and humans in the interface areas of the Serengeti ecosystem. Transmission can be either from wild animals to livestock and vice versa, from wild animals to livestock then to humans or from wild animals directly to humans [17, 20]. Indeed it has been reported earlier, that wild animals can act as a source of infection for livestock and humans [34, 35].
Wildebeest migrate seasonally from the Serengeti to the Maasai Mara for pastures, a practice likely to spread Brucella in the Serengeti ecosystem [36]. Zebra constantly intermingle with wildebeest during grazing, living together in close association. This behavior could be the basis for the transmission of pathogens amongst wild animals [33, 37].
In this study, qPCR was observed to have a higher detection rate of Brucella spp. than AMOS PCR. This finding is supported by reports from other studies which compared the two platforms and reported qPCR as superior tool [29, 38, 39]. Most likely because the quantitative PCR is more sensitive to lower concentrations of DNA than conventional methods [29, 40]. It could however, also depend on the biotypes of Brucella circulating in the region, for example B. abortus biovar 3 which has previously been detected in Tanzania cannot be detected using the AMOS PCR [18, 41].
The AMOS PCR is designed to detect B. abouts, B. melitensis, B. ovis and B. suis, while the qPCR used in the present study was able to differentiate B. abortus and B. melitensis [29, 30, 42]. A positive result was considered only if a sample was positive by both assays. B. suis was detected in the AMOS PCR but could not be confirmed by the qPCR assay used. Future studies could build on these findings to conduct further molecular studies in wildlife samples, using more advanced typing techniques like the Bruce ladder [43] or multi-locus sequence analysis [41, 44].
This study had a number of limitations; first, the samples used in this study were collected on a convenience/availability basis and the study was not systematically designed to determine epidemiological inference for respective animal species. Secondly, although the qPCR assays sensitively detected Brucella DNA in these archived samples, we did not have sufficient quantities and quality of genomic DNA to confirm the species and sub-types using more advanced typing techniques. Lastly, although the study exploited molecular techniques on DNA extracted directly from clinical samples, archived for up to 15 years, most of the source material was of inadequate quality to perform serological testing or confirmatory culture. Future studies could target more freshly collected samples and explore options for immunological and bacteriology confirmatory tests in this critical yet under-studied population.
Conclusions
Findings from this study show the robust use of molecular techniques for the detection of Brucella in DNA extracted directly from archived wild animal field samples. This has great potential in expanding the detection of brucellosis among populations that may be hard to reach or sample, and particularly in wild animals, where sample collection is expensive, dangerous and tedious. Numerous wildlife management and research institutions however, have samples in archive from previous field activities. This study has shown that there are Brucella spp. circulating in different wild animal species in the Serengeti ecosystem. Most of Brucella spp. detected have zoonotic potential. Detection of zoonotic Brucella species in wild animals suggests that livestock and humans at the interface areas are at risk of acquiring the infection, underscoring the need for a One Health approach for the control of this disease. The findings from this study, although contextual to the Serengeti ecosystem, provide valuable insights into Brucella infection and host associations in wild animal population applicable to much of sub-Saharan Africa.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Abbreviations
- AMOS PCR:
-
Multiplex polymerase chain reaction for Brucella abortus, Brucella melitensis,Brucella ovis and Brucella suis
- DNA:
-
Dioxyribonucleic acid
- EDTA:
-
Ethylenediaminetetraacetic acid
- Kb:
-
Kilobyte
- PCR:
-
Polymerase chain reaction
- qPCR:
-
Quantitative Real-Time PCR
- SUA:
-
Sokoine University of Agriculture
- TAWIRI:
-
Tanzania Wildlife Research Institute
- WHO:
-
World Health Organization
References
Pappas G, Papadimitriou P, Akritidis N, Christou L, Tsianos EV. The new global map of human brucellosis. Lancet Infect Dis. 2006;6:91–9. https://doi.org/10.1016/S1473-3099(06)70382-6.
Moreno E. Retrospective and prospective perspectives on zoonotic brucellosis. Front Microbiol. 2014;5:1–18. https://doi.org/10.3389/fmicb.2014.00213.
Arimi SM, Koroti E, Kang’ethe EK, Omore AO, McDermott JJ. Risk of infection with Brucella abortus and Escherichia coli O157:H7 associated with marketing of unpasteurized milk in Kenya. Acta Trop. 2005;96:1–8. https://doi.org/10.1016/j.actatropica.2005.05.012.
Mantur BG, Amarnath SK. Brucellosis in India - A review. J Biosci. 2008;33:539–47. https://doi.org/10.1007/s12038-008-0072-1.
Matope G, Bhebhe E, Muma JB, Oloya J, Madekurozwa RL, Lund A, et al. Seroprevalence of brucellosis and its associated risk factors in cattle from smallholder dairy farms in Zimbabwe. Trop Anim Health Prod. 2011;43:975–82. https://doi.org/10.1007/s11250-011-9794-4.
World Health Organization (WHO). Integrated control of neglected zoonotic diseases in Africa: Applying the “One Health” concept. Nairobi: WHO; 2009. https://hdl.handle.net/10568/3378.
Davis SD, Templeton WJ, Ficht AT, Williams DJ, Kopec DJ, Garry A. Brucella abortus in Captive Bison; Serology, Bacteriology, Pathogenesis and Transmission to Cattle. J Wildl Dis. 1990;26:360–71. https://doi.org/10.7589/0090-3558-26.3.360.
Bishop GC, Bosman PP, Herr S. Bovine brucellosis. In: Coetzer JAW, Thomson GR, Tustin RC, editors. Infectious Diseases of Livestock with Special Reference to Southern Africa II. Cape Town: Oxford University press, UK; 1994. p. 1053–66.
Waghela S, Karstad L. Antibodies to Brucella spp. among blue wildebeest and African buffalo in Kenya. J Wildl Dis. 1986;22:189–92. https://doi.org/10.7589/0090-3558-22.2.189.
Madsen M, Anderson EC. Serologic Survey of Zimbabwean Wildlife for Brucellosis. J Zoo Wildl Med. 1995;26:240–245. Available: http://www.jstor.org/stable/20095468.
Hamblin C, Anderson EC, Hirji K, Jago M, Mlengeya T. Antibodies to some pathogenic agents in free-living wild species in Tanzania. Epidemiol Infect. 1990;105:585–94. https://doi.org/10.1017/S0950268800048226.
Fyumagwa RD, Wambura PN, Mellau LSB, Hoare R. Seroprevalence of Brucella abortus in buffaloes and wildebeests in the Serengeti ecosystem: A threat to humans and domestic ruminants. Tanzania Vet J. 2010;26:62–7. https://doi.org/10.4314/tvj.v26i2.53803.
Mellau LSB, Kuya SL, Wambura PN. Seroprevalence of brucellosis in domestic ruminants in livestock-wildlife interface: A case study of Ngorongoro Conservation Area, Arusha, Tanzania. Tanzania Vet J. 2009;26:44–50. https://doi.org/10.4314/tvj.v26i1.49234.
Temba PB, Mwakapuja RS, Makondo ZE, Ndaki KL, Mdegela RH, Moser I, et al. Spatial distribution and risk factors for brucellosis in domestic and wild animals at livestock-wildlife interface in Mikumi-Selous ecosystem, Tanzania. Tanzania Vet J. 2019;34:1–8. Available: https://www.ajol.info/index.php/tvj/article/view/188479.
Sachs R, Staak C, Groocock CM. Serological investigation of brucellosis in game animals in Tanzania. Bull Epizoot Dis Afr. 1968;16:93–100. Available: http://europepmc.org/MED/5693879.
The United Republic of Tanzania (URT). National Strategy for Prevention and Control of Brucellosis in Humans and Animals 2018–2023. Prime Minist Off. 2018. https://doi.org/10.1111/fcre.12333.
Assenga JA, Matemba LE, Muller SK, Malakalinga JJ, Kazwala RR. Epidemiology of Brucella infection in the human, livestock and wildlife interface in the Katavi-Rukwa ecosystem, Tanzania. BMC Vet Res. 2015;11:1–11. https://doi.org/10.1186/s12917-015-0504-8.
Mathew C, Stokstad M, Johansen TB, Klevar S, Mdegela RH, Mwamengele G, et al. genotypic characterization of Brucella abortus First isolation, identification, phenotypic and genotypic characterization of Brucella abortus biovar 3 from dairy cattle in Tanzania. BMC Vet Res. 2015;11:9. https://doi.org/10.1186/s12917-015-0476-8.
Gwida M, Al-Dahouk S, Melzer F, Rösler U. Brucellosis - Regionally Emerging Zoonotic Disease? Croat Med J. 2010;51:289–95. https://doi.org/10.3325/cmj.2010.51.289.
Ducrotoy M, Bertu WJ, Matope G, Cadmus S, Conde-Álvarez R, Gusi AM, et al. Brucellosis in Sub-Saharan Africa: Current challenges for management, diagnosis and control. Acta Trop. 2017;165:179–93. https://doi.org/10.1016/j.actatropica.2015.10.023.
United Republic of Tanzania (URT), United States Department of Defense (DoD), Defense Threat Reduction Agency (DTRA), Cooperative Threat Reduction (CTR), Cooperative Biological Engagement Program (CBEP). The United Republic of Tanzania One Health Strategic Plan 2015–2020. Dar es Salaam; 2015. Available: http://www.tzdpg.or.tz/fileadmin/documents/dpg_internal/dpg_working_groups_clusters/cluster_2/health/Key_Sector_Documents/Tanzania_Key_Health_Documents/FINAL_URT_One_Health_Strategy_Plan_20151021.pdf.
Shirima GM, Kunda JS. Prevalence of brucellosis in the human, livestock and wildlife interface areas of Serengeti National Park, Tanzania. Onderstepoort J Vet Res. 2016;83:2–5. https://doi.org/10.4102/ojvr.v83i1.1032.
Pastoralists Indigenous Non-Governmental Organization’s Forum (PINGO). Socio-Economic Contributions of Pastoralism as a Livelihood System in Tanzania: Case of Selected Pastoral Districts in Arusha, Manyara and Dar Es Salaam Regions. Arusha, Tanzania; 2016. doi:https://www.pingosforum.or.tz/index.php/about-us/reports/studies/53-socio-economic-study/file.
Makala R, Majigo MV, Bwire GM, Kibwana U, Mirambo MM, Joachim A. Seroprevalence of Brucella infection and associated factors among pregnant women receiving antenatal care around human, wildlife and livestock interface in Ngorongoro ecosystem, Northern Tanzania. A cross-sectional study. BMC Infect Dis. 2020;20:1–7. https://doi.org/10.1186/s12879-020-4873-7.
Bodenham FR, Lukambagire SA, Ashford R, Buza JJ, Cash-Goldwasser S, Crump AJ, et al. Prevalence and speciation of acute brucellosis in febrile patients from a pastoralist community of Tanzania. Sci Rep. 2020;10:1–10. https://doi.org/10.1038/s41598-020-62849-4.
Mkiramweni NP, DeLacy T, Jiang M, Chiwanga FE. Climate change risks on protected areas ecotourism: shocks and stressors perspectives in Ngorongoro Conservation Area, Tanzania. J Ecotourism. 2016;15:139–57. https://doi.org/10.1080/14724049.2016.1153645.
Navarro E, Casao MA, Solera J. Diagnosis of human brucellosis using PCR. Expert Rev Mol Diagn. 2004;115–23. https://doi.org/10.1586/14737159.4.1.115.
Bricker BJ, Halling SM. Differentiation of Brucella abortus bv. 1, 2, and 4, Brucella melitensis, Brucella ovis, and Brucella suis bv. 1 by PCR. J Clin Microbiol. 1994;32:2660–6. https://doi.org/10.1128/jcm.32.11.2660-2666.1994.
Matero P, Hemmilä H, Tomaso H, Piiparinen H, Rantakokko-Jalava K, Nuotio L, et al. Rapid field detection assays for Bacillus anthracis, Brucella spp., Francisella tularensis and Yersinia pestis. Clin Microbiol Infect. 2011;17:34–43. https://doi.org/10.1111/j.1469-0691.2010.03178.x.
Probert WS, Schrader KN, Khuong NY, Bystrom SL, Graves MH. Real-Time Multiplex PCR Assay for Detection of Brucella spp., B. abortus, and B. melitensis. J Clin Microbiol. 2004;42:1290–3. https://doi.org/10.1128/JCM.42.3.1290-1293.2004.
R Core Team. R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2020. Available: https://www.r-project.org/.
Zerva L, Bourantas K, Mitka S, Kansouzidou A. Serum Is the Preferred Clinical Specimen for Diagnosis of Human Brucellosis by PCR. J Clin Microbiol. 2001;39:1661–4. https://doi.org/10.1128/JCM.39.4.1661.
Godfroid J. Brucellosis in wildlife. OIE Rev Sci Tech. 2002;21:277–86. https://doi.org/10.20506/rst.21.2.1333.
Mathias LA, Girio RJS, Duarte JMB. Serosurvey for antibodies against Brucella abortus and Leptospira interrogans in pampas deer from Brazil. J Wildl Dis. 1999;35:112–4. https://doi.org/10.7589/0090-3558-35.1.112.
Muma JB, Samui KL, Oloya J, Munyeme M, Skjerve E. Risk factors for brucellosis in indigenous cattle reared in livestock–wildlife interface areas of Zambia. Prev Vet Med. 2007;80:306–17. https://doi.org/10.1016/J.PREVETMED.2007.03.003.
McNaughton SJ. Serengeti migratory wildebeest: Facilitation of energy flow by grazing. Science. 1976;191:92–4. https://doi.org/10.1126/science.191.4222.92.
Stoffregen WC, Olsen SC, Wheeler CJ, Bricker BJ, Palmer MV, Jensen AE, et al. Diagnostic Characterization of a Feral Swine Herd Enzootically Infected with Brucella. J Vet Diagnostic Investig. 2007;19:227–37. https://doi.org/10.1177/104063870701900301.
Pérez-sancho M, García-seco T, Arrogante L, García N, Martínez I, Diez-guerrier A. Research in Veterinary Science Development and evaluation of an IS 711 -based loop mediated isothermal amplification method (LAMP) for detection of Brucella spp. on clinical samples. 2013. https://doi.org/10.1016/j.rvsc.2013.05.002.
Wareth G, Melzer F, Tomaso H, Roesler U, Neubauer H. Detection of Brucella abortus DNA in aborted goats and sheep in Egypt by real-time PCR Veterinary Research. BMC Res Notes. 2015;8:1–5. https://doi.org/10.1186/s13104-015-1173-1.
Al-Dahouk S, Sprague LD, Neubauer H. New developments in the diagnostic procedures for zoonotic brucellosis in humans. Sci Tech Rev Off Int des Epizoot. 2013;32:177–88. https://doi.org/10.20506/rst.32.1.2204.
Whatmore AM, Koylass MS, Muchowski J, Edwards-Smallbone J, Gopaul KK, Perrett LL. Extended multilocus sequence analysis to describe the global population structure of the genus Brucella: Phylogeography and relationship to biovars. Front Microbiol. 2016;7:1–14. https://doi.org/10.3389/fmicb.2016.02049.
Baily GG, Krahn BJ, Drasar BS, Stocker NG. Detection of Brucella melitensis and Brucella abortus by DNA amplification. J Trop Med Hyg. 1992;95: 271–275. Available: http://www.ajtmh.org/archives.
Jamil T, Melzer F, Njeru J, El-Adawy H, Neubauer H, Wareth G. Brucella abortus: Current Research and Future Trends. Curr Clin Microbiol Rep. 2017;4:1–10. https://doi.org/10.1007/s40588-017-0052-z.
Whatmore AM, Perrett LL, MacMillan AP. Characterisation of the genetic diversity of Brucella by multilocus sequencing. BMC Microbiol. 2007;7:1–15. https://doi.org/10.1186/1471-2180-7-34.
Acknowledgements
SRM, CM, AL, JA and RBY acknowledge support from the DELTAS Africa Initiative [Afrique One-ASPIRE /DEL-15-008]. Afrique One-ASPIRE is funded by a consortium of donor including the African Academy of Sciences (AAS) Alliance for Accelerating Excellence in Science in Africa (AESA), the New Partnership for Africa’s Development Planning and Coordinating (NEPAD) Agency, the Wellcome Trust [107753/A/15/Z] and the UK government.
We gratefully acknowledge laboratory technicians at the Sokoine University of Agriculture, and College of Veterinary of Medicine and Biomedical Sciences and Kilimanjaro Clinical Research Institute-Biotechnology Laboratories for technical assistance, Tanzania Wildlife Research Institute (TAWIRI) for their willingness in sharing the samples.
Funding
This project is supported within the framework of the DELTAS Africa Initiative [Afrique One-ASPIRE /DEL-15-008]. Afrique One-ASPIRE is funded by a consortium of donor including the African Academy of Sciences (AAS) Alliance for Accelerating Excellence in Science in Africa (AESA), the New Partnership for Africa’s Development Planning and Coordinating (NEPAD) Agency, the Wellcome Trust [107753/A/15/Z] and the UK government. The funders had no role in the design of the study, data collection, analysis, interpretation, writing or decision on the choice for the journal.
Author information
Authors and Affiliations
Contributions
Concept development: Bonfoh B., Kazwala R. R, Fokou G. Funding acquisition: Bonfoh B., Kazwala R. R, Fokou G. Investigation and formal analysis: Sambu, R. M, Nonga, H.E., Mathew C., Lukambagire A. S., Akoko J. Methodology: Sambu, R. M, Mathew, C., Lukambagire (A) S., Akoko J., Richard (B) Yapi, Kazwala R. R. Supervision: Nonga, H.E., Mathew C, Keyyu J.D., Kazwala R. R. Writing original draft: Sambu, R. M., Lukambagire A.S., Mathew (C) Writing, review & editing: All authors. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was conducted in conformity with the ethical and animal welfare guidelines stipulated by Sokoine University of Agriculture research regulations and guidelines of 2019. A research permit was provided by the Tanzania Wildlife Research Institute (TAWIRI), for study conduct on wildlife (TWRI/RS/57/VOL IV/85/72).
Consent to participate is not applicable in this study.
Consent for publication
Not applicable
Competing interests
The authors declare no conflict of interest. The findings and conclusions in this paper are those of the authors and do not necessarily represent the official position of the participating institutions or the funding organization.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary materials S1.
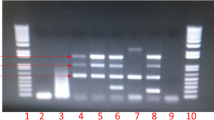
Brucella spp. detected by AMOS PCR from wildlife in the Serengeti ecosystem. The first and the last lanes are 10kb DNA ladder, lanes 2- 12, are positive samples, lanes 13 and 14 are negative samples, lane NC is a negative control containing nuclease free water and lane PC is a positive control comprising DNA of B. abortus strain RB51
Additional file 2: Supplementary materials S2.
A data set with results from the three PCR assays (AMOS PCR, Brucella genus specific qPCR and qPCR speciation) performed.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
Cite this article
Sambu, R.M., Mathew, C., Nonga, H.E. et al. Circulating Brucella species in wild animals of the Serengeti ecosystem, Tanzania. One Health Outlook 3, 15 (2021). https://doi.org/10.1186/s42522-021-00047-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42522-021-00047-6