
Abstract
Background
Hypothalamic hamartoma (HH) is a congenital non-progressive lesion of hypothalamus during fetal development. Mass-like lesions in different anatomical locations often develop a variously disabling course presenting with cognitive decline, psychiatric symptoms, as well as multiple seizure types. As a rare disease, HH is relatively common in infants and children, but it is extremely rare in adults.
Case presentation
We reported a case of adult-onset hypothalamic hamartoma, and summarized and analyzed relevant reports and studies of HH worldwide. The patient had clinical manifestations characterized by multiple seizure forms. After stereotactic radiofrequency thermocoagulation and drug treatment, the condition was effectively controlled. The patient was followed up till October 2022, with no recurrence of seizures.
Conclusions
Epilepsy caused by HH can resemble that of temporal lobe seizures, as HH forms a complex epileptogenic network with other regions of the brain through anatomical and functional connections. Early treatment of HH can provide better control of the symptoms of epilepsy, and patients with longer disease courses may have more complications.
Similar content being viewed by others


Background
Hypothalamic hamartoma (HH) is an ectopic, non-neoplastic nerve tissue containing normal neurons and glial cells (oligodendrocytes and astrocytes) [1], which generally originates from the third ventricular floor, tuber cinereum, or mammillary body [2, 3]. HH is extremely rare in clinic, with an incidence of one per 50,000–100,000 [4]. HH can cause neurological and endocrine symptoms, including gelastic seizure (GS), dacrystic seizure, and other types of epilepsy. It can also lead to precocious puberty, abnormal behavior, and progressive cognitive deterioration [1]. Although HH is common in infants and children, it is extremely rare in adults [5, 6]. Nguyen reviewed 277 HH patients published in literature and found that the average age of onset of HH was 2.49 years [5]. Among 214 HH patients reported by Beijing Tiantan Hospital, only 2.3% developed HH in adulthood [7]. In the present study, we retrospectively review the clinical characteristics of an adult HH patient without GS, who was treated in the Department of Neurology of our hospital. We will also provide a review of previous studies on adult HH.
Case presentation
Clinical Data
A 24-year-old female patient was admitted to our hospital for "paroxysmal loss of consciousness over more than 1 year". In April 2020, the patient developed episodic unconsciousness, first deja vu, fear, and palpitations, followed by unconsciousness accompanied by vocalization, lip smacking, straightening of the left upper limb, involuntary groping of the right hand, and wandering around, which lasted for 2–3 minutes each time, and occurred 2–3 times a week. She had received treatment at other hospitals and had been diagnosed with "neurosis". The patient’s condition did not improve after treatment with "Deanxit and escitalopram oxalate". In mid-March 2021, a new type of seizure appeared, manifested as a dazed condition, slanting of the right corner of the mouth, loss of consciousness, and uplifting of the right upper limb. Thereafter, the head, neck, and body deflected to the right, and she fell to the ground slowly, with the upper limbs being straight and rigid for half a minute. The seizure attacks occurred three times a week on average. She was admitted to our hospital on April 25, 2021. The neurological examination showed no obvious abnormality. Cranial magnetic resonance imaging (MRI) showed a HH on the left (Fig. 1). Video electroencephalogram (EEG) monitoring showed abnormal waveforms during interictal periods (Fig. 2). During the monitoring period, she had one seizure attack, characterized by accelerated heart rate, lifting and stiffening of the right upper limb, loss of consciousness, lip smacking, involuntary groping with the left upper limb, and a pedal-like movement of the right lower limb. EEG manifestations during this seizure are presented in Fig. 3.

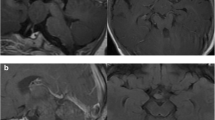
Cranial magnetic resonance imaging (MRI). Axial T2WI (a), T1WI (b), coronal T2WI (c), and T2 FLAIR (d) all showed presence of HH in the left side, with equal signal shadows. The HH had a size of 9 mm * 5 mm, and protruded into the third ventricle

EEG in the interictal period, indicating abnormal discharges in the bilateral temporal areas and sphenoid electrode area(blue arrow), significant on the left side

EEG recordings during a seizure. The EEG traces showed fast-wave activity in bilateral temporal areas, significant on the left, and then evolved into sharp-wave rhythms in the left temporal area, frontal pole, and frontal area, and finally spread to the whole lead, showing slow-wave rhythm distribution
After discussion with the clinical team, the patient’s diagnosis was confirmed to be symptomatic epilepsy and HH. The primary seizures were focal with disturbances of consciousness, leading to secondary generalized seizures. In May 2021, the patient was treated with stereotactic radiofrequency thermocoagulation (SRT) at a maximum temperature of 60–65 °C for 60 s, followed by treatment with 1 g of levetiracetam twice per day and 300 mg of oxcarbazepine twice per day after the operation. The patient experienced no seizures during the year after the surgery. In June 2022, the patient stopped taking the drug on her own, and no seizures were observed until October 2022. The postoperative cranial MRI is shown in Fig. 4.

Postoperative cranial MRI T1WI (a) and T2WI (b)
Discussion
The case presented in our report experienced a variety of seizure forms that appeared in adulthood, manifested mainly as focal seizures accompanied by disturbance of consciousness and secondary generalized seizures. The patient had no history of GS. This is rarely seen as most HH patients have epilepsy in the form of GS or have a history of GS [8, 9]. A recent study reported that most HH patients without GS would have a very late-onset age [10]. Sone et al. [11] also reported a female case of adult-onset HH with a disease course of eight years, presenting as intractable epilepsy without GS. In our case, clinical manifestations and EEG examination suggested that the seizures may originate from the temporal lobe. However, brain MRI examination revealed a left-sided HH with a diameter of 9 mm, and no abnormal signals were observed in the left temporal lobe.
A similarity between the case reported by Sone et al. and our case is that both cases showed clinical manifestations and video EEG activity that suggested origination of the seizures from the temporal lobe. To clarify the origination of seizures, Sone et al. [11] using stereotactic intracranial EEG (SEEG) recordings and found that the ictal discharges from the HH preceded those from other regions including the left hippocampus. In our case, although the seizure was not confirmed to originate from the HH by SEEG, it was effectively controlled after destruction of the HH by SRT. So why did these patients show symptoms similar to temporal lobe seizures? Ictal single-photon emission computerized tomography studies have demonstrated the presence of increased functional connectivity (FC) between the HH/hypothalamic region and the ipsilateral thalamus, especially with the anterior thalamic nucleus (a relay to the anterior cingulate and limbic circuit) and the thalamic nucleus (a relay to the orbitofrontal, anterior cingulate, and medial premotor, as well as the parietal cortical areas) [12,13,14]. HH is thus able to form a complex epileptogenic network over the range of the HH through various anatomical and functional connections [10, 15]. Kahane et al. [16] reported the complete Grenoble series of 5 cases showing gelastic or dacrystic seizures correlated with discharges in the HH, although other types of seizures were related to discharges affecting the neocortical regions. The HH might work as a pacemaker, and the seizure activities of the HH are generated by the intrinsic pacemaker neurons [17]. The seizure activity can spread to adjacent cortical structures such as the frontal and temporal lobes, which then act as secondary foci of epileptogenesis [18]. Two major phenotypes can be distinguished based on electroclinical features: (i) focal seizures with epigastric or dejà-vu auras, loss of consciousness and oroalimentary or gestural automatisms suggestive of temporal lobe involvement; and (ii) motor seizures with tonic, atonic, myoclonic, or versive phenomena, suggesting frontoparietal network involvement [19]. Thus, in our patient, the seizures of similar temporal origin were caused by HH triggering of temporal neocortical discharges.
The differences between the two cases lie in the course and prognosis. The patient reported by Sone et al. had a disease course of eight years, and took medications of "sodium valproate, carbamazepine and lamotrigine", but still experienced complex partial seizures approximately once every 2-3 months. The patient was then treated with SRT, and only took lamotrigine after the operation. Although the seizures did not recur after three years of follow-up, the patient’s cognitive function did not recover. In our report, the case had a disease course of one year, with seizures being effectively controlled in the early stage of the disease without cognitive impairment. The seizure activities of the HH are usually present as drug-resistant epilepsy, but as the epilepsy intrinsically arises within the hamartoma, ablation of the hamartoma by any method can result in remission of the seizure activity [13, 20, 21]. Schulze-Bonhage et al. [22] also found that early discovery, diagnosis, and treatment offer better control of epileptic symptoms. Treatment at the late stages is associated with a lower success rate and a higher incidence of complications such as cognitive impairment. A longer duration of epilepsy is also been related to the poorer prognosis after HH-targeted surgical procedures. In the Strasbourg-Kork series, 80% of patients with epilepsy duration less than 10 years achieved complete seizure freedom, whereas only 20% of those with disease for over 20 years achieved seizure-free [23]. These results indicate that early diagnosis and early treatment are of great significance in improving the prognosis of HH patients.
Conclusions
In conclusion, HH without gelastic seizures is easily misdiagnosed as it may occur in a form similar to temporal lobe seizures. Early treatment of HH leads to improved control of epileptic symptoms and patients with a longer course of disease may have more complications. Therefore, early diagnosis and treatment is of great significance to improve the prognosis of HH patients.
Availability of data and materials
Not applicable.
Abbreviations
- EEG:
-
Electroencephalogram
- GS:
-
Gelastic seizure
- HH:
-
Hypothalamic hamartoma
- MRI:
-
Magnetic Resonance Imaging
- PP:
-
Precocious puberty
- SEEG:
-
Stereotactic intracranial Electroencephalogram
- SRT:
-
Stereotactic Radiofrequency Thermocoagulation
References
Alomari SO, Houshiemy MNE, Bsat S, Moussalem CK, Allouh M, Omeis IA. Hypothalamic hamartomas: A comprehensive review of the literature - Part 1: Neurobiological features, clinical presentations and advancements in diagnostic tools. Clin Neurol Neurosurg. 2020;197:106076.
Coons SW, Rekate HL, Prenger EC, Wang N, Drees C, Ng YT, et al. The histopathology of hypothalamic hamartomas: study of 57 cases. J Neuropathol Exp Neurol. 2007;66(2):131–41.
Freeman JL, Harvey AS, Rosenfeld JV, Wrennall JA, Bailey CA, Berkovic SF. Generalized epilepsy in hypothalamic hamartoma: evolution and postoperative resolution. Neurology. 2003;60(5):762–7.
Khawaja AM, Pati S, Ng YT. Management of Epilepsy Due to Hypothalamic Hamartomas. Pediatr Neurol. 2017;75:29–42.
Nguyen D, Singh S, Zaatreh M, Novotny E, Levy S, Testa F, et al. Hypothalamic hamartomas: seven cases and review of the literature. Epilepsy Behav. 2003;4(3):246–58.
Craig DW, Itty A, Panganiban C, Szelinger S, Kruer MC, Sekar A, et al. Identification of somatic chromosomal abnormalities in hypothalamic hamartoma tissue at the GLI3 locus. Am J Hum Genet. 2008;82(2):366–74.
Li D. Clinical characteristics of 214 cases of hypothalamic hamartoma. Chin J Neurosurg. 2009;06:497–9.
Kameyama S, Shirozu H, Masuda H. Asymmetric gelastic seizure as a lateralizing sign in patients with hypothalamic hamartoma. Epilepsy Behav. 2019;94:35–40.
Kameyama S, Shirozu H, Masuda H, Ito Y, Sonoda M, Akazawa K. MRI-guided stereotactic radiofrequency thermocoagulation for 100 hypothalamic hamartomas. J Neurosurg. 2016;124(5):1503–12.
Liu C, Hu W, Zhang C, Zheng Z, Yang X, Wang X, et al. Anatomical features decide the atypical seizure manifestation of parahypothalamic hamartomas. Front Neurol. 2022;13:981488. https://doi.org/10.3389/fneur.2022.981488.
Sone D, Kaido T, Watanabe M, Murata Y, Taniguchi G, Otsuki T. Adult-onset refractory epilepsy with hypothalamic hamartoma and no gelastic seizures successfully treated by stereotactic thermocoagulation: A case report. Seizure. 2016;37:32–4.
Valentin A, Lazaro M, Mullatti N, Cervantes S, Malik I, Selway RP, et al. Cingulate epileptogenesis in hypothalamic hamartoma. Epilepsia. 2011;52(5):e35–9.
Conde Blanco E, Anciones Martín C, Manzanares I, Gil López F, Roldán P, Donaire A, et al. Hypothalamic hamartomas in adulthood: Clinical spectrum and treatment outcome-A unicenter experience. Brain Behav. 2019;9(11):e01412.
Kameyama S, Masuda H, Murakami H. Ictogenesis and symptomatogenesis of gelastic seizures in hypothalamic hamartomas: an ictal SPECT study. Epilepsia. 2010;51(11):2270–9.
Scholly J, Bartolomei F. Gelastic seizures and the hypothalamic hamartoma syndrome: Epileptogenesis beyond the lesion? Handb Clin Neurol. 2021;182:143–54.
Kahane P, Ryvlin P, Hoffmann D, Minotti L, Benabid AL. From hypothalamic hamartoma to cortex: what can be learnt from depth recordings and stimulation? Epileptic Disord. 2003;5(4):205–17.
Liu Z, Luan G, Yang C, Guan Y, Liu C, Wang J, et al. Distinguishing Dependent-Stage Secondary Epileptogenesis in a Complex Case of Giant Hypothalamic Hamartoma With Assistance of a Computational Method. Front Neurol. 2020;11:478.
Silva AHD, Lo WB, Mundil NR, Walsh AR. Transtemporal approach to hypothalamic hamartomas in children: report of 3 cases [published online ahead of print, 2020 Feb 28]. J Neurosurg Pediatr. 2020:1–9. https://doi.org/10.3171/2019.12.PEDS19231.
Chibbaro S, Cebula H, Scholly J, Todeschi J, Ollivier I, Timofeev A, et al. Pure endoscopic management of epileptogenic hypothalamic hamartomas. Neurosurg Rev. 2017;40(4):647–53.
Qasim BA, Mohammed AA. Hamartoma of hypothalamus presented as precocious puberty and epilepsy in a 10-year-old girl. Int J Surg Case Rep. 2020;77:170–3.
Cohen NT, Cross JH, Arzimanoglou A, Berkovic SF, Kerrigan JF, Miller IP, et al. Hypothalamic Hamartomas: Evolving Understanding and Management. Neurology. 2021;97(18):864–73.
Schulze-Bonhage A, Trippel M, Wagner K, Bast T, Deimling FV, Ebner A, et al. Outcome and predictors of interstitial radiosurgery in the treatment of gelastic epilepsy. Neurology. 2008;71(4):277–82.
Scholly J, Staack AM, Kahane P, Scavarda D, Régis J, Hirsch E, et al. Hypothalamic hamartoma: Epileptogenesis beyond the lesion? Epilepsia. 2017;58(Suppl 2):32–40.
Acknowledgements
This study was partly supported by the General Hospital of Southern Theatre Command PLA, China.
Funding
This study was supported by the foundation of Medical Science and Technology of Guangdong (A2022402).
Author information
Authors and Affiliations
Contributions
WH and CJ conceived the study and wrote the manuscript. BD and ZL supervised the analyses and study. ZQ, YZ and JL collected and organized the datasets. ZL and WX analyzed and interpreted the EEG results. All authors have participated in the discussion, examinations, and/or management of the patients. WH and CJ contributed equally to this work. The author(s) read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The patient and her family had provided informed consent. This case report has been exempt from the ethics approval by the Ethics Committee of the General Hospital of Southern Theater Command.
Consent for publication
The patient agreed to publish this article and has signed an informed consent form.
Competing interests
The authors declare no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Han, W., Jiang, C., Qi, Z. et al. Adult-onset hypothalamic hamartoma: origin of epilepsy?. Acta Epileptologica 5, 11 (2023). https://doi.org/10.1186/s42494-023-00120-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42494-023-00120-9