
Abstract
The ketogenic diet (KD) is a high-fat, low-carbohydrate diet, in which fat is used as the primary energy source through the production of ketone bodies (KBs) in place of glucose. The KD was formally introduced in 1921 to mimic the biochemical changes associated with fasting and gained recognition as a potent treatment for pediatric epilepsy in the mid-1990s. The clinical and basic scientific knowledge that supports the anti-seizure efficacy, safety, and feasibility of using the KD in patients with epilepsy is huge. Additionally, the International Ketogenic Diet Study Group’s consensus guidelines provide practical information in 2009 and 2018. The KD is a broad-spectrum therapy for drug resistant epilepsy and is gaining attention as a potential therapy for other neurological disorders. This article will review recent aspects on the use of the KD, including its mechanisms of action, KD alternatives, expanding its use across different age groups and regions, its use as a treatment for other neurologic disorders, and future research subjects.
Similar content being viewed by others
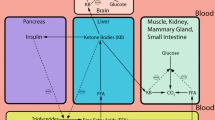
History of the ketogenic diet
The ketogenic diet (KD) is a high-fat, low-carbohydrate diet in which fat is used as the primary energy source through the formation of ketone bodies (KBs) in place of glucose [1]. The use of fasting in patients with epilepsy was well recorded (from at least 500 BC) and is noted as a treatment for epilepsy in the Hippocratic writing [2]. The KD was formally introduced in 1921 to mimic the biochemical changes associated with fasting and gained recognition as a potent treatment for pediatric epilepsy in the mid-1990s [3, 4]. The clinical and basic scientific knowledge that supports the anti-seizure efficacy, safety, and feasibility of using the KD in patients with epilepsy is huge. Additionally, the International Ketogenic Diet Study Group’s consensus guidelines provide practical information on patient selection, pre-KD counseling, diet selection and implementation, dietary supplementation, follow-up evaluation, adverse effects, and discontinuation of the diet [5, 6]. Currently, the KD should be considered in patients after two anti-epileptic drugs (AEDs) have failed; its application might be earlier for specific epilepsy and genetic syndromes. The KD has been established as one of four main treatments for drug-resistant epilepsy (DRE), together with new AEDs, surgery, and neuromodulation. Compared with surgical treatment, the use of the KD is reversible, inexpensive, and easy to access.
The introduction of KD alternatives, such as the use of medium-chain triglycerides (MCTs), the modified Atkin’s diet (MAD), and the low glycemic index treatment (LGIT), made ketogenic therapy less complicated and more accessible [6]. The KD has been expanded to specific age groups such as infants and adults and introduced in many countries, including resource-limited regions worldwide [7,8,9]. Although preliminary, the KD has also been used to treat various medical illnesses, like as autism spectrum disorder (ASD), Alzheimer’s disease (AD), traumatic brain injury, and brain tumors [10,11,12].
This article will review recent aspects on the use of the KD, including its mechanisms of action, KD alternatives, expanding its use across different age groups and regions, its use as a treatment for other neurologic disorders, and future research subjects.
Mechanisms of action of the ketogenic diet
Over the past two decades, there has been a steady increase in knowledge about the underlying mechanisms of the KD [13,14,15]. The understanding of these mechanisms is incomplete, but multiple mechanisms are likely to be interconnected. Such pathways involving neuronal excitability, energy metabolism, and neuroprotection form the basis of many candidate mechanisms. The clinical experiences of patients can be associated with these mechanisms and applied to such diseases as mitochondrial disorders, febrile infection-related epilepsy syndrome (FIRES), and neurodegenerative disorders such as AD and Parkinson’s disease; such associations have motivated new research studies into the mechanisms of action of the KD [16,17,18,19,20,21,22].
Early clinical and research studies of the anti-seizure mechanism of the KD have focused on KBs produced in the liver via fatty acid metabolism. However, both clinical and preclinical studies have shown inconsistent results between the diet’s anti-seizure effects and KB levels [23]. These observations suggest that other various mechanisms play roles.
One of the anti-seizure mechanisms of KD is to inhibit the brain by increasing γ-aminobutyric acid (GABA). Usually, glutamate is converted into either GABA or aspartate, but KD depletes oxaloacetate, which is needed to convert glutamate into aspartate. As a result, more GABA from glutamate in CSF was generated [24]. Adenosine triphosphate (ATP)-sensitive potassium channels are activated by the KD, leading to membrane hyperpolarization that prevents neuronal excitability and directly inhibits vesicular glutamate transport, thereby decreasing the amount of glutamate present during synaptic transmission and providing positive feedback to GABAergic channels [25,26,27].
Chronic ketosis has a positive impact on mitochondrial biogenesis and metabolism by increasing the brain’s energy reserves via a reduction in the excitability of synapses, resulting in elevated levels of mitochondrial ATP production [28]. That increased ATP leads to an increased level of adenosine, which generates antiepileptic activity through the adenosine receptors in the brain by inhibiting the glutaminergic system or opening ATP-sensitive potassium channels [24]. Further, when the patient receives a high-fat diet, it promotes the oxidation of fatty acids and alters polyunsaturated fatty acids (PUFAs) in the body. PUFAs are thought to protect neurons by the stimulation of mitochondrial uncoupling protein. Thus, the KD causes an increase in PUFAs, which ultimately reduces the production of reactive oxygen species. PUFAs can directly regulate different types of ion pumps and channels; It blocks Na+ and Ca++ channels and opens K+ channels, thereby reducing hyperexcitability [29].
It is well acknowledged that inflammatory mechanisms contribute to the pathophysiology of chronic epilepsy and neurodegenerative disorders [30,31,32,33]. Controlling inflammation in chronic epilepsy is essential, as shown in the treatment with steroid-responsive infantile epileptic encephalopathy and KD in FIRES [20, 34]. In addition, preclinical and clinical studies on inflammatory mechanisms in neurodegeneration are also continuing [12, 35, 36]. Anti-inflammatory effects by altering both pro-inflammatory and anti-inflammatory mediators, reduced interleukin 1β, and other pro-inflammatory cytokines in peripheral blood and brain tissues of rats fed with a KD, was noted [37]. However, these inflammatory mediators fluctuate greatly subject to the tissue types and the time of measurement [32].
There is increased interest in identifying the link between the gut and the brain, and the effects of diet on gut microbiota appear to be significant [38,39,40]. In terms of gut microbiota and KD-related anti-seizure properties, dysbiosis can enhance susceptibility for epilepsy and accelerate illness resulting from chronic restraint stress [41]. Olson et al. proved the impact of gut microbiota on the antiseizure effects of the KD with two mouse models, showing an increase in beneficial Akkermansia and Parabacteroides bacteria and a decrease in bacterial diversity. Furthermore, this microbiota transformation brings about changes in the colonic luminal metabolome, with decreases in gamma-glutamylated amino acids in the blood, resulting in increased GABA/glutamate levels in the brain. These modifications in neurotransmitters’ expression might contribute to the antiseizure effect of the KD [42]. Children with DRE showed evidence of a reduction in the abundance and diversity of gut microbiota, and there are differences in the composition of gut microbiota between the responder and non-responder groups after 6 months on the KD [43].
Alternatives and modification of the ketogenic diet
The term “ketogenic diet” describes to any diet therapy in which the dietary composition brings about a ketogenic stage of metabolism. Following the development of the classic KD, which consists of long-chain triglycerides (LCTs), usually applied as a ratio of fat to nonfat (protein and carbohydrate) as 4:1 or 3:1, new alternative diets have been proposed to increase retention and palatability, while mimicking the effects produced by the classic KD. There are currently four forms of KDs: the classic KD, the MCT diet, the MAD, and the LGIT [6].
As for the MCT diet, in which fats are provided through triglycerides comprising about 60% octanoic acid and 40% decanoic acid. It yields more ketones per kilocalorie from the rapid metabolism of the shorter fatty acids compared to LCTs [44, 45]. This increased ketogenic potential signifies that less total fat is required in the MCT diet, allowing for more carbohydrate and protein consumption and broader food choices. In addition, studies have indicated that medium-chain fatty acids provided in the MCT diet could directly inhibit AMPA receptors and enhance mitochondrial function [46, 47]. With these mechanisms, the MCT diet helps in reduction of seizure occurrence and plays positive roles in other neurological disorders, such as AD, cancer, and diabetes [45].
The MAD consists of a 1:1 to 1.5:1 ketogenic ratio, with no restriction on protein, fluids, or calories, making the diet more tolerable with easier meal planning [48]. This diet’s primary purpose is for children who have behavioral difficulties and are reluctant to consume the classic KD. The LGIT involves exchanging high glycemic index (GI) foods (e.g., most refined carbohydrates) with low GI alternatives (e.g., meat, dairy, some unprocessed whole-grain foods, and some fresh fruits and vegetables) [49]. The GI is defined as the tendency of food to raise blood sugar compared to an equivalent amount of a reference carbohydrate. While the LGIT does not necessarily produce a condition of ketosis, both a decrease in glucose metabolism and constant glucose levels play roles in the mechanisms of action of the LGIT. Therefore, these various diet options, such as the MAD and LGIT, could be suitable for adolescents and adults and initiated on an outpatient basis or used with limited resources, such as dietitians in resource-limited lesions.
The efficacy of these three KD alternatives has been compared to that of the classic KD, including in three randomized controlled trials. While each treatment’s efficacy depends on the patient’s age, specific epilepsy syndrome, and etiology, these alternatives are not inferior to classic KD treatments [50,51,52].
If it is necessary to maintain a KD for several years or more due to seizure recurrence or the clinical course of the disease, it may be practical to switch to the MAD or LGIT and discontinue the KD when considering the risks of long-term complications such as growth and cardiovascular changes [6].
Expanding the use of the ketogenic diet
Although the knowledge related to the expansion of indications related to specific epilepsy syndromes is essential, there is also a need to expand the age range for which the diet is used and provide information on the KD to more people around the world to allow better access to its benefits [9, 53].
The first 2 years of life are critical for rapid brain growth and psychosocial development related to synapse formation and myelination [54]. Such young patients are most at risk for neurodevelopmental compromises over time. Additionally, the incidence of epilepsy is highest during this period, and KBs constitute a significant energy source for brain development in utero and infancy [55, 56]. Thus, active and safe use of KD at this age is recommended.
At first, the KD was not used in infants, as it was considered that this age group could not maintain a state of ketosis. However, the clinical efficacy and experience of the KD in this age group have been accumulating, including application in newborns and in cases with early onset developmental and epileptic encephalopathy (DEE) [57,58,59,60]. A systemic and meta-analysis of KD usage for infants published in 2020 and updated guidelines in 2018 by the International Ketogenic Diet Study Group reveal that KD treatment responses are confirmed to be higher in infants than in other age groups without much concern for side effects; Approximately 33% of infants on the KD became seizure-free, with 59% achieving a greater than 50% reduction in seizures [6, 61]. Also, specific guidelines for infants were created in 2016 [62]. Additional benefits of KD on cognition and behavior have been reported [61, 63]. However, these results are lacking consistency, and further studies are warranted. KD’s use is expanding again by applying MAD and LGIT to adolescents and adults, and these age groups are previously regarded unsuitable for dietary treatment due to lack of compliance [64, 65]. Therefore, the KD’s method used in the most challenging way in a limited group is shifting to a more straightforward approach for broader age groups.
Although the widespread use of the KD, the large regions of the world such as the Caribbean, Central America, Africa, Eastern Europe, and Southeast Asia do not offer this treatment [9, 66,67,68]. Therefore, encouraging the use of the KD in more countries and regions is needed. In addition, the rate of intractable epilepsy is about 20–30% of patients diagnosed with epilepsy [69]. While such patients may benefit from new drugs, surgery, and neuromodulation, these options are nearly impossible due to the cost and lack of supplies in resource-deficient countries. Compared to the methods mentioned above, the use of the KD is much more achievable in these areas, and the approach has been made easier due to the development of KD alternatives during the last 20 years. For this purpose, the International League Against Epilepsy published a special report in 2015, listing the minimum requirements for KD in resource-limited regions and providing KD workshops for healthcare providers in areas like Indonesia, Mongolia, and Uzbekistan [9].
Use of the ketogenic diet for other neurological disorders
The systemic action of the KD can elicit a broad spectrum of effects, and there has been interest in applying the KD for neurological disorders other than epilepsy.
Although the data are preliminary, the KD has been used for many neurological disorders, such as ASD, AD, brain tumors, and traumatic brain injury [10, 11]. Specifically, interest in neurodegenerative disorders has arisen through studies of neuroprotection and energy metabolism related to the KD’s mechanisms [11, 70].
The updated guideline in 2018 newly included specific genetic syndromes and epilepsy, such as FIRES and Angelman syndrome, in the KD beneficial group, which means the efficacy is higher (> 70%) than the average efficacy of KD [6, 71]. Through a retrospective review of 23 cases, the LGIT provides Angelman syndrome patients with a high level of seizure control with minimal side effects [71]. In accordance with 2015 systematic review, eight studies (five human and three animal) studied the effects of the KD (the classic KD or MAD) in ASD, and the classic KD was evaluated as being the most favorable non-AED treatment for improving seizures; it also showed beneficial effects on other relevant clinical factors connected to ASD [72]. For example, a prospective pilot study showed that 18 out of 30 children with autism scored better on an autism rating scale after consuming a KD, and a case report noted that a girl with both autism and epilepsy improved from severely autistic to non-autistic while on a KD [73, 74].
KBs are viewed as primary mediators to prevent aging and neurodegeneration in AD via enhanced mitochondrial functioning [11]. Impaired brain glucose metabolism and amyloid-β plaques may lead to AD development, and the KD likely reduces the formation of these plaques and replaces glucose with alternative energy sources in the brain [21]. There are also reports that medium chain fatty acid of the MCT diet plays a more critical role than KBs itself in AD [45].
The potential anti-tumor effects of the KD are also of growing interest. Previous studies have suggested that the KD inhibits tumor cell growth by changing cellular metabolism, which is likely to increase responses to other anti-tumor treatments [75, 76]. A case study of two glioma patients reported that the treatment of the KD alone seemed ineffective for impeding tumor growth but had promising effects when combined with standard treatments, which may be explained by the control of primary brain tumor progression by an energy-restricted KD [76].
New research subjects
In general, the KD has been shown to improve seizure outcomes in 50% of patients but offers no benefit to the remaining patients. Predicting KD efficacy is an important subject; due to studies on predictive factors, such as age, etiology, and specific epilepsy syndromes, KD indications for groups with excellent results have been identified [77, 78]. These predictors used to rely on clinical phenotypes, but with the recent advances of genetic testing technologies, such as next-generation sequencing, physicians now can diagnose various genetic etiologies of epilepsy [79, 80]. Therefore, KD efficacy prediction based on etiology might be necessary. Ko et al. compared the efficacy of the KD according to specific gene mutations, discovering that the use of the KD is effective in patients with SCN1A, KCNQ2, STXBP1, and SCN2A mutations but ineffective to the patients with CDKL5 mutations [57]. Other case series and international collaborating experiences in DEE have been continuously reported [60, 81]. Thus, with genetic improvements, the etiology-based identification for selecting better candidates will be possible in the future.
Metabolic epilepsies occur in relation to rare inborn errors of metabolism (IEM). One of the representative clinical manifestations of certain neurological disorders, such as rare IEM or genetic diseases in infancy, is refractory epilepsy. Although the number of IEM is rare, its collective prevalence is 1 in 1000, and most have no cure. Given that the KD is a primary metabolism-based treatment that brings about a broad range of biochemical, hormonal, and physiological effects, it is also possible to consider its use as a treatment for IEM [82].
The KD is the treatment of choice for two distinct disorders of brain energy disorder: glucose transporter type 1 deficiency syndrome and pyruvate dehydrogenase deficiency [83, 84]. The KD supplies ketones that bypass these metabolic defects and act as an alternative energy source in the brain. The effects of the KD have been reported for other metabolic disorders, such as those of mitochondria. Clinical findings have demonstrated that the KD may effectively treat children with DRE associated with mitochondrial respiratory chain complex defects and mitochondrial encephalopathy with lactic acidosis and stroke-like episodes [17, 18, 85]. Before these clinical results were published, the KD was generally thought of as a contraindicated treatment for mitochondrial disorders. Since the KD mechanisms were shown to enhance mitochondrial function and biogenesis, the KD is now indicated and confirmed to be effective in treating mitochondrial disorders, especially complex 1 deficiency syndrome. Although single reports, the use of KD in other metabolic diseases, such as phosphofructokinase deficiency and adenylosuccinate lyase deficiency, were listed [86, 87].
The mechanisms of action of the KD in other IEM remain unclear. The diet’s multiple mechanisms appear to counter the negative consequences of genetic mutations [82]. Moreover, its beneficial effects can be broadly categorized into the following groups: the restoration of impaired bioenergetics and synaptic dysfunction, improvements in redox homeostasis, and enhancements in its anti-inflammatory and epigenetic activities. Thus, it is important to try carefully to pay attention to its contraindications (e.g., in fatty acid oxidation disorders).
Conclusion
The KD, which was initially established as a non-pharmacological but powerful treatment for patients with epilepsy, reemerged in the mid-1990s in a variety of forms. Since then, numerous mechanisms of the KD have been proposed. Its use has been expanded to various neurological disorders other than DRE. Also, its use has been proved positive for broader age groups and patients with DEE or IEM, which started to become identifiable with the recent genetic diagnostic technology. With all these proven benefits, we hope to see more of its use in resource-limited regions. Regardless of the type of disease, age, more people are expected to benefit from the KD.
Availability of data and materials
Not applicable.
Abbreviations
- KD:
-
Ketogenic diet
- KBs:
-
Ketone bodies
- AEDs:
-
Anti-epileptic drugs
- DRE:
-
Drug-resistant epilepsy
- MCTs:
-
Medium-chain triglycerides
- MAD:
-
Modified Atkin’s diet
- LGIT:
-
Low glycemic index treatment
- ASD:
-
Autism spectrum disorder
- AD:
-
Alzheimer’s disease
- FIRES:
-
Febrile infection-related epilepsy syndrome
- GABA:
-
γ-aminobutyric acid
- ATP:
-
Adenosine triphosphate
- PUFAs:
-
Polyunsaturated fatty acids
- LCTs:
-
Long-chain triglycerides
- GI:
-
Glycemic index
- DEE:
-
Developmental and epileptic encephalopathy
- IEM:
-
Inborn errors of metabolism
References
Koppel SJ, Swerdlow RH. Neuroketotherapeutics: a modern review of a century-old therapy. Neurochem Int. 2018;117:114–25. https://doi.org/10.1016/j.neuint.2017.05.019.
Wheless JW. History of the ketogenic diet. Epilepsia. 2008;49(Suppl 8):3–5. https://doi.org/10.1111/j.1528-1167.2008.01821.x.
Wilder RM. The effects of ketonemia on the course of epilepsy. May Clin Proc. 1921;2:307–8.
Bailey EE, Pfeifer HH, Thiele EA. The use of diet in the treatment of epilepsy. Epilepsy Behav. 2005;6(1):4–8. https://doi.org/10.1016/j.yebeh.2004.10.006.
Kossoff EH, Zupec-Kania BA, Amark PE, Ballaban-Gil KR, Christina Bergqvist AG, Blackford R, et al. Optimal clinical management of children receiving the ketogenic diet: recommendations of the international ketogenic diet study group. Epilepsia. 2009;50(2):304–17. https://doi.org/10.1111/j.1528-1167.2008.01765.x.
Kossoff EH, Zupec-Kania BA, Auvin S, Ballaban-Gil KR, Christina Bergqvist AG, Blackford R, et al. Optimal clinical management of children receiving dietary therapies for epilepsy: updated recommendations of the International Ketogenic Diet Study Group. Epilepsia Open. 2018;3(2):175–92. https://doi.org/10.1002/epi4.12225.
Nordli DR Jr, Kuroda MM, Carroll J, Koenigsberger DY, Hirsch LJ, Bruner HJ, et al. Experience with the ketogenic diet in infants. Pediatrics. 2001;108(1):129–33. https://doi.org/10.1542/peds.108.1.129.
Klein P, Tyrlikova I, Mathews GC. Dietary treatment in adults with refractory epilepsy: a review. Neurology. 2014;83(21):1978–85. https://doi.org/10.1212/WNL.0000000000001004.
Kossoff EH, Al-Macki N, Cervenka MC, Kim HD, Liao J, Megaw K, et al. What are the minimum requirements for ketogenic diet services in resource-limited regions? Recommendations from the International League Against Epilepsy Task Force for Dietary Therapy. Epilepsia. 2015;56(9):1337–42. https://doi.org/10.1111/epi.13039.
Barañano KW, Hartman AL. The ketogenic diet: uses in epilepsy and other neurologic illnesses. Curr Treat Options Neurol. 2008;10(6):410–9. https://doi.org/10.1007/s11940-008-0043-8.
Stafstrom CE, Rho JM. The ketogenic diet as a treatment paradigm for diverse neurological disorders. Front Pharmacol. 2012;3:59.
Rusek M, Pluta R, Ulamek-Koziol M, Czuczwar SJ. Ketogenic diet in Alzheimer's disease. Int J Mol Sci. 2019;20(16):3892. https://doi.org/10.3390/ijms20163892.
Youngson NA, Morris MJ, Ballard JWO. The mechanisms mediating the antiepileptic effects of the ketogenic diet, and potential opportunities for improvement with metabolism-altering drugs. Seizure. 2017;52:15–9. https://doi.org/10.1016/j.seizure.2017.09.005.
Rogawski MA, Löscher W, Rho JM. Mechanisms of action of antiseizure drugs and the ketogenic diet. Cold Spring Harb Perspect Med. 2016;6(5). https://doi.org/10.1101/cshperspect.a022780.
Alqahtani F, Imran I, Pervaiz H, Ashraf W, Perveen N, Rasool MF, et al. Non-pharmacological interventions for intractable epilepsy. Saudi Pharm J. 2020;28(8):951–62. https://doi.org/10.1016/j.jsps.2020.06.016.
Gano LB, Patel M, Rho JM. Ketogenic diets, mitochondria, and neurological diseases. J Lipid Res. 2014;55(11):2211–28. https://doi.org/10.1194/jlr.R048975.
Lee YM, Kang HC, Lee JS, Kim SH, Kim EY, Lee SK, et al. Mitochondrial respiratory chain defects: underlying etiology in various epileptic conditions. Epilepsia. 2008;49(4):685–90. https://doi.org/10.1111/j.1528-1167.2007.01522.x.
Kang HC, Lee YM, Kim HD, Lee JS, Slama A. Safe and effective use of the ketogenic diet in children with epilepsy and mitochondrial respiratory chain complex defects. Epilepsia. 2007;48(1):82–8. https://doi.org/10.1111/j.1528-1167.2006.00906.x.
Paleologou E, Ismayilova N, Kinali M. Use of the ketogenic diet to treat intractable epilepsy in mitochondrial disorders. J Clin Med. 2017;6(6):56. https://doi.org/10.3390/jcm6060056..
Nabbout R, Mazzuca M, Hubert P, Peudennier S, Allaire C, Flurin V, et al. Efficacy of ketogenic diet in severe refractory status epilepticus initiating fever induced refractory epileptic encephalopathy in school age children (FIRES). Epilepsia. 2010;51(10):2033–7. https://doi.org/10.1111/j.1528-1167.2010.02703.x.
Broom GM, Shaw IC, Rucklidge JJ. The ketogenic diet as a potential treatment and prevention strategy for Alzheimer's disease. Nutrition. 2019;60:118–21. https://doi.org/10.1016/j.nut.2018.10.003.
Vanitallie TB, Nonas C, Di Rocco A, Boyar K, Hyams K, Heymsfield SB. Treatment of Parkinson disease with diet-induced hyperketonemia: a feasibility study. Neurology. 2005;64(4):728–30. https://doi.org/10.1212/01.WNL.0000152046.11390.45.
Simeone TA, Simeone KA, Rho JM. Ketone Bodies as Anti-Seizure Agents. Neurochem Res. 2017;42(7):2011–8. https://doi.org/10.1007/s11064-017-2253-5.
Yudkoff M, Daikhin Y, Nissim I, Horyn O, Lazarow A, Luhovyy B, et al. Response of brain amino acid metabolism to ketosis. Neurochem Int. 2005;47(1–2):119–28. https://doi.org/10.1016/j.neuint.2005.04.014.
Lutas A, Yellen G. The ketogenic diet: metabolic influences on brain excitability and epilepsy. Trends Neurosci. 2013;36(1):32–40. https://doi.org/10.1016/j.tins.2012.11.005.
Juge N, Gray JA, Omote H, Miyaji T, Inoue T, Hara C, et al. Metabolic control of vesicular glutamate transport and release. Neuron. 2010;68(1):99–112. https://doi.org/10.1016/j.neuron.2010.09.002.
D'Andrea Meira I, Romao TT, Pires do Prado HJ, Kruger LT, MEP P, da Conceicao PO. Ketogenic diet and epilepsy: what we know so far. Front Neurosci. 2019;13:5.
Bough KJ, Wetherington J, Hassel B, Pare JF, Gawryluk JW, Greene JG, et al. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann Neurol. 2006;60(2):223–35. https://doi.org/10.1002/ana.20899.
Bough KJ, Rho JM. Anticonvulsant mechanisms of the ketogenic diet. Epilepsia. 2007;48(1):43–58. https://doi.org/10.1111/j.1528-1167.2007.00915.x.
Chamorro A, Hallenbeck J. The harms and benefits of inflammatory and immune responses in vascular disease. Stroke. 2006;37(2):291–3. https://doi.org/10.1161/01.STR.0000200561.69611.f8.
Vezzani A, Granata T. Brain inflammation in epilepsy: experimental and clinical evidence. Epilepsia. 2005;46(11):1724–43. https://doi.org/10.1111/j.1528-1167.2005.00298.x.
Vezzani A, Friedman A. Brain inflammation as a biomarker in epilepsy. Biomark Med. 2011;5(5):607–14. https://doi.org/10.2217/bmm.11.61.
Walker L, Sills GJ. Inflammation and epilepsy: the foundations for a new therapeutic approach in epilepsy? Epilepsy Curr. 2012;12(1):8–12. https://doi.org/10.5698/1535-7511-12.1.8.
Vigevano F, Arzimanoglou A, Plouin P, Specchio N. Therapeutic approach to epileptic encephalopathies. Epilepsia. 2013;54(Suppl 8):45–50. https://doi.org/10.1111/epi.12423.
Lilamand M, Mouton-Liger F, Paquet C. Ketogenic diet therapy in Alzheimer's disease: an updated review. Curr Opin Clin Nutr Metab Care. 2021;24(4):372–8. https://doi.org/10.1097/MCO.0000000000000759.
French JA, Koepp M, Naegelin Y, Vigevano F, Auvin S, Rho JM, et al. Clinical studies and anti-inflammatory mechanisms of treatments. Epilepsia. 2017;58(Suppl 3):69–82. https://doi.org/10.1111/epi.13779.
Dupuis N, Curatolo N, Benoist JF, Auvin S. Ketogenic diet exhibits anti-inflammatory properties. Epilepsia. 2015;56(7):e95–8. https://doi.org/10.1111/epi.13038.
David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505(7484):559–63. https://doi.org/10.1038/nature12820.
Peng A, Qiu X, Lai W, Li W, Zhang L, Zhu X, et al. Altered composition of the gut microbiome in patients with drug-resistant epilepsy. Epilepsy Res. 2018;147:102–7. https://doi.org/10.1016/j.eplepsyres.2018.09.013.
Chatzikonstantinou S, Gioula G, Kimiskidis VK, McKenna J, Mavroudis I, Kazis D. The gut microbiome in drug-resistant epilepsy. Epilepsia Open. 2021;6(1):28–37. https://doi.org/10.1002/epi4.12461.
Medel-Matus JS, Shin D, Dorfman E, Sankar R, Mazarati A. Facilitation of kindling epileptogenesis by chronic stress may be mediated by intestinal microbiome. Epilepsia Open. 2018;3(2):290–4. https://doi.org/10.1002/epi4.12114.
Olson CA, Vuong HE, Yano JM, Liang QY, Nusbaum DJ, Hsiao EY. The gut microbiota mediates the anti-seizure effects of the ketogenic diet. Cell. 2018;173(7):1728–41.e13.
Zhang Y, Zhou S, Zhou Y, Yu L, Zhang L, Wang Y. Altered gut microbiome composition in children with refractory epilepsy after ketogenic diet. Epilepsy Res. 2018;145:163–8. https://doi.org/10.1016/j.eplepsyres.2018.06.015.
Huttenlocher PR, Wilbourn AJ, Signore JM. Medium-chain triglycerides as a therapy for intractable childhood epilepsy. Neurology. 1971;21(11):1097–103. https://doi.org/10.1212/WNL.21.11.1097.
Augustin K, Khabbush A, Williams S, Eaton S, Orford M, Cross JH, et al. Mechanisms of action for the medium-chain triglyceride ketogenic diet in neurological and metabolic disorders. Lancet Neurol. 2018;17(1):84–93. https://doi.org/10.1016/S1474-4422(17)30408-8.
Chang P, Terbach N, Plant N, Chen PE, Walker MC, Williams RS. Seizure control by ketogenic diet-associated medium chain fatty acids. Neuropharmacology. 2013;69:105–14. https://doi.org/10.1016/j.neuropharm.2012.11.004.
Hughes SD, Kanabus M, Anderson G, Hargreaves IP, Rutherford T, O'Donnell M, et al. The ketogenic diet component decanoic acid increases mitochondrial citrate synthase and complex I activity in neuronal cells. J Neurochem. 2014;129(3):426–33. https://doi.org/10.1111/jnc.12646.
Kossoff EH, McGrogan JR, Bluml RM, Pillas DJ, Rubenstein JE, Vining EP. A modified Atkins diet is effective for the treatment of intractable pediatric epilepsy. Epilepsia. 2006;47(2):421–4. https://doi.org/10.1111/j.1528-1167.2006.00438.x.
Pfeifer HH, Thiele EA. Low-glycemic-index treatment: a liberalized ketogenic diet for treatment of intractable epilepsy. Neurology. 2005;65(11):1810–2. https://doi.org/10.1212/01.wnl.0000187071.24292.9e.
Kim JA, Yoon JR, Lee EJ, Lee JS, Kim JT, Kim HD, et al. Efficacy of the classic ketogenic and the modified Atkins diets in refractory childhood epilepsy. Epilepsia. 2016;57(1):51–8. https://doi.org/10.1111/epi.13256.
Neal EG, Chaffe H, Schwartz RH, Lawson MS, Edwards N, Fitzsimmons G, et al. A randomized trial of classical and medium-chain triglyceride ketogenic diets in the treatment of childhood epilepsy. Epilepsia. 2009;50(5):1109–17. https://doi.org/10.1111/j.1528-1167.2008.01870.x.
Wells J, Swaminathan A, Paseka J, Hanson C. Efficacy and safety of a ketogenic diet in children and adolescents with refractory epilepsy-A review. Nutrients. 2020;12(6):1809. https://doi.org/10.3390/nu12061809.
Armeno M, Caraballo R. The evolving indications of KD therapy. Epilepsy Res. 2020;163:106340. https://doi.org/10.1016/j.eplepsyres.2020.106340.
Dobbing J, Sands J. Quantitative growth and development of human brain. Arch Dis Child. 1973;48(10):757–67. https://doi.org/10.1136/adc.48.10.757.
Eltze CM, Chong WK, Cox T, Whitney A, Cortina-Borja M, Chin RF, et al. A population-based study of newly diagnosed epilepsy in infants. Epilepsia. 2013;54(3):437–45. https://doi.org/10.1111/epi.12046.
Cotter DG, d'Avignon DA, Wentz AE, Weber ML, Crawford PA. Obligate role for ketone body oxidation in neonatal metabolic homeostasis. J Biol Chem. 2011;286(9):6902–10. https://doi.org/10.1074/jbc.M110.192369.
Ko A, Jung DE, Kim SH, Kang HC, Lee JS, Lee ST, et al. The efficacy of ketogenic diet for specific genetic mutation in developmental and epileptic encephalopathy. Front Neurol. 2018;9:530. https://doi.org/10.3389/fneur.2018.00530.
Thompson L, Fecske E, Salim M, Hall A. Use of the ketogenic diet in the neonatal intensive care unit-safety and tolerability. Epilepsia. 2017;58(2):e36–e9. https://doi.org/10.1111/epi.13650.
Le Pichon JB, Thompson L, Gustafson M, Abdelmoity A. Initiating the ketogenic diet in infants with treatment refractory epilepsy while maintaining a breast milk diet. Seizure. 2019;69:41–3. https://doi.org/10.1016/j.seizure.2019.03.017.
Lim Z, Wong K, Olson HE, Bergin AM, Downs J, Leonard H. Use of the ketogenic diet to manage refractory epilepsy in CDKL5 disorder: experience of >100 patients. Epilepsia. 2017;58(8):1415–22. https://doi.org/10.1111/epi.13813.
Lyons L, Schoeler NE, Langan D, Cross JH. Use of ketogenic diet therapy in infants with epilepsy: a systematic review and meta-analysis. Epilepsia. 2020;61(6):1261–81. https://doi.org/10.1111/epi.16543.
van der Louw E, van den Hurk D, Neal E, Leiendecker B, Fitzsimmon G, Dority L, et al. Ketogenic diet guidelines for infants with refractory epilepsy. Eur J Paediatr Neurol. 2016;20(6):798–809. https://doi.org/10.1016/j.ejpn.2016.07.009.
van Berkel AA, DM IJ, Verkuyl JM. Cognitive benefits of the ketogenic diet in patients with epilepsy: a systematic overview. Epilepsy Behav. 2018;87:69–77. https://doi.org/10.1016/j.yebeh.2018.06.004.
Nei M, Ngo L, Sirven JI, Sperling MR. Ketogenic diet in adolescents and adults with epilepsy. Seizure. 2014;23(6):439–42. https://doi.org/10.1016/j.seizure.2014.02.015.
McDonald TJW, Cervenka MC. Lessons learned from recent clinical trials of ketogenic diet therapies in adults. Curr Opin Clin Nutr Metab Care. 2019;22(6):418–24. https://doi.org/10.1097/MCO.0000000000000596.
Seo JH, Kim HD. Cultural challenges in using the ketogenic diet in Asian countries. Epilepsia. 2008;49(Suppl 8):50–2. https://doi.org/10.1111/j.1528-1167.2008.01834.x.
Kossoff EH, McGrogan JR. Worldwide use of the ketogenic diet. Epilepsia. 2005;46(2):280–9. https://doi.org/10.1111/j.0013-9580.2005.42704.x.
Cao D, Badoe E, Zhu Y, Zhao X, Hu Y, Liao J. First application of ketogenic diet on a child with intractable epilepsy in Ghana. Child Neurol Open. 2015;2(3):2329048X15604593.
Berg AT. Identification of pharmacoresistant epilepsy. Neurol Clin. 2009;27(4):1003–13. https://doi.org/10.1016/j.ncl.2009.06.001.
Gasior M, Rogawski MA, Hartman AL. Neuroprotective and disease-modifying effects of the ketogenic diet. Behav Pharmacol. 2006;17(5–6):431–9. https://doi.org/10.1097/00008877-200609000-00009.
Grocott OR, Herrington KS, Pfeifer HH, Thiele EA, Thibert RL. Low glycemic index treatment for seizure control in Angelman syndrome: a case series from the Center for Dietary Therapy of epilepsy at the Massachusetts General Hospital. Epilepsy Behav. 2017;68:45–50. https://doi.org/10.1016/j.yebeh.2016.12.018.
Castro K, Faccioli LS, Baronio D, Gottfried C, Perry IS, dos Santos Riesgo R. Effect of a ketogenic diet on autism spectrum disorder: a systematic review. Res Autism Spectr Disord. 2015;20:31–8. https://doi.org/10.1016/j.rasd.2015.08.005.
Evangeliou A, Vlachonikolis I, Mihailidou H, Spilioti M, Skarpalezou A, Makaronas N, et al. Application of a ketogenic diet in children with autistic behavior: pilot study. J Child Neurol. 2003;18(2):113–8. https://doi.org/10.1177/08830738030180020501.
Herbert MR, Buckley JA. Autism and dietary therapy: case report and review of the literature. J Child Neurol. 2013;28(8):975–82. https://doi.org/10.1177/0883073813488668.
Klement RJ, Champ CE, Otto C, Kämmerer U. Anti-tumor effects of ketogenic diets in mice: a meta-analysis. PLoS One. 2016;11(5):e0155050. https://doi.org/10.1371/journal.pone.0155050.
Schwartz K, Chang HT, Nikolai M, Pernicone J, Rhee S, Olson K, et al. Treatment of glioma patients with ketogenic diets: report of two cases treated with an IRB-approved energy-restricted ketogenic diet protocol and review of the literature. Cancer Metab. 2015;3(1):3. https://doi.org/10.1186/s40170-015-0129-1.
Prezioso G, Carlone G, Zaccara G, Verrotti A. Efficacy of ketogenic diet for infantile spasms: a systematic review. Acta Neurol Scand. 2018;137(1):4–11. https://doi.org/10.1111/ane.12830.
Cross JH. The ketogenic diet in the treatment of Lennox-Gastaut syndrome. Dev Med Child Neurol. 2012;54(5):394–5. https://doi.org/10.1111/j.1469-8749.2012.04276.x.
Møller RS, Dahl HA, Helbig I. The contribution of next generation sequencing to epilepsy genetics. Expert Rev Mol Diagn. 2015;15(12):1531–8. https://doi.org/10.1586/14737159.2015.1113132.
Dunn P, Albury CL, Maksemous N, Benton MC, Sutherland HG, Smith RA, et al. Next generation sequencing methods for diagnosis of epilepsy syndromes. Front Genet. 2018;9:20. https://doi.org/10.3389/fgene.2018.00020.
Tian X, Zhang Y, Zhang J, Lu Y, Men X, Wang X. Ketogenic diet in infants with early-onset epileptic encephalopathy and SCN2A mutation. Yonsei Med J. 2021;62(4):370–3. https://doi.org/10.3349/ymj.2021.62.4.370.
Gavrilovici C, Rho JM. Metabolic epilepsies amenable to ketogenic therapies: indications, contraindications, and underlying mechanisms. J Inherit Metab Dis. 2021;44(1):42–53. https://doi.org/10.1002/jimd.12283.
Klepper J, Leiendecker B. GLUT1 deficiency syndrome--2007 update. Dev Med Child Neurol. 2007;49(9):707–16. https://doi.org/10.1111/j.1469-8749.2007.00707.x.
Sofou K, Dahlin M, Hallböök T, Lindefeldt M, Viggedal G, Darin N. Ketogenic diet in pyruvate dehydrogenase complex deficiency: short- and long-term outcomes. J Inherit Metab Dis. 2017;40(2):237–45. https://doi.org/10.1007/s10545-016-0011-5.
Steriade C, Andrade DM, Faghfoury H, Tarnopolsky MA, Tai P. Mitochondrial encephalopathy with lactic acidosis and stroke-like episodes (MELAS) may respond to adjunctive ketogenic diet. Pediatr Neurol. 2014;50(5):498–502. https://doi.org/10.1016/j.pediatrneurol.2014.01.009.
Swoboda KJ, Specht L, Jones HR, Shapiro F, DiMauro S, Korson M. Infantile phosphofructokinase deficiency with arthrogryposis: clinical benefit of a ketogenic diet. J Pediatr. 1997;131(6):932–4. https://doi.org/10.1016/S0022-3476(97)70048-9.
Jurecka A, Opoka-Winiarska V, Rokicki D, Tylki-Szymańska A. Neurologic presentation, diagnostics, and therapeutic insights in a severe case of adenylosuccinate lyase deficiency. J Child Neurol. 2012;27(5):645–9. https://doi.org/10.1177/0883073811424465.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
Hye Eun Kwon conceive and draft the manuscript. Heung Dong Kim reviewed and edited the manuscript before submission. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
Author Heung Dong Kim is a member of the Editorial Board for Acta Epileptologica. Author Heung Dong Kim was not involved in the journal’s review of, or decisions related to this manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kwon, H.E., Kim, H.D. Recent aspects of ketogenic diet in neurological disorders. Acta Epileptologica 3, 21 (2021). https://doi.org/10.1186/s42494-021-00053-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42494-021-00053-1