
Abstract
Picornaviridae are a family of small positive-strand RNA viruses, and transmitted via the respiratory or fecal-oral route. The neurotropic picornaviruses can induce acute or late recurrent seizures following central nervous system infection, by infecting the peripheral nerve, crossing the blood-brain barrier and migrating in the Trojan-horse method. Theiler’s murine encephalomyelitis virus (TMEV), as a member of Picornaviridae family, can cause encephalitis, leading to chronic spontaneous seizures. TMEV-infected C57BL/6 mice have been used as an animal model for exploring the mechanism of epileptogenesis and assessing new antiepileptic drugs. Astrogliosis, neuronal death and microglial recruitment have been detected in the hippocampus following the picornaviruse-induced encephalitis. The macrophages, monocytes, neutrophils, as well as IL-6 and TNF-α released by them, play an important role in the epileptogenesis. In this review, we summarize the clinical characteristics of picornavirus infection, and the immunopathology involved in the TMEV-induced epilepsy.
Similar content being viewed by others
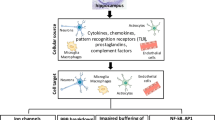


Background
Epilepsy is one of the most common neurological disorders that affects over 70 million people worldwide [1] and leads to 13 million disability adjusted life years each year. Epilepsy can also cause cognitive and psychosocial impairment [2]. According to the new classification of epilepsies by the International League Against Epilepsy in 2017, the etiology of epilepsy can be structural, genetic, infectious, metabolic, immune or unknown [3]. Emerging evidence has demonstrated that the viral infection plays a role in the epileptogenesis, especially in developing countries [4, 5]. The incidence of acute seizures in encephalitis patients following the infection of neurotropic viruses varies from 7 to 67%, depending on the type of virus [6]. Besides, the status epilepticus in the acute stage of viral encephalitis can be life-threatening [7]. Furthermore, patients who survived from viral encephalitis are 16 times more likely to develop late unprovoked seizures [8,9,10]. Currently, there is a lack of sufficient evidence for the use of anticonvulsant prophylaxis for acute and late seizures in viral encephalitis [11]. Thus, intensive investigation of the mechanisms of seizure induced by viral infection may provide insights into the prevention of seizures following viral encephalitis.
To date, over 100 viruses have been reported to be associated with epileptogenesis, such as human herpes virus type 6, influenza A viruse, rotavirus and picornaviruses [12]. As one of the largest viral families, Picornaviridae contains 54 species of small positive-strand RNA viruses, and is transmitted though the respiratory or fecal-oral route [13]. Some of the picornaviruses are neurotropic and can cause fatal encephalitis and meningoencephalitis concurrent with seizures [14, 15]. For animals, C57BL/6 mice infected by the Theiler’s murine encephalomyelitis virus (TMEV) have been shown to suffer recurrent seizures following polioencephalitis, and can be used as an animal model to explore the underlying mechanisms of epileptogenesis [16,17,18]. In this review, we will summarize the clinical characteristics of seizures induced by picornaviruses and the mechanisms of epileptogenesis triggered by TMEV infection.
Picornaviruses that induce seizures
Rhinoviruses, enteroviruses and parechovirus are picornaviruses commonly detected to induce encephalitis with seizures [19,20,21,22]. Neurotropic picornaviruses can infect the peripheral nerve, cross the blood-brain barrier (BBB) and migrate in the Trojan-horse manner, thereby infecting the central nervous system (CNS) [14, 23,24,25].
Human rhinovirus (HRV) is a common cause of respiratory infection. Seizures can occur in 22.7% of patients with HRV infection, which is higher than the other viruses in the respiratory tract [26]. Seizures mostly occur in patients with underlying neurological abnormalities or a history of seizure [26], although the mechanisms remain unknown. However, several causes for the seizures in the context of HRV infection have been proposed. For example, cytokines, such as interleukin-6 and interleukin-1, are related to the development of seizures [27]. The direct viral infection may also promote this process.
Human parechoviruses (HPeVs) belong to the Parechovirus genus of the Picornaviridae family [28]. HPeV infections are often associated with gastrointestinal and respiratory symptoms, but the encephalitis caused by HPeV3 with concomitant seizures have been reported in infants. In most patients, the development of seizure outbreak is followed by repeated seizures with an average interval of 9.5 h between the first and last seizures [29]. Central apnea and focal neurological signs have been described in some young infants with seizures [21]. The initial seizure symptom is often accompanied by multiple white matter lesions [30].
Coxsackievirus and other enteroviruses are the most possible cause of myocarditis. Epidemiological evidence has shown that infants with coxsackievirus infection present with high mortality, while young adults may have a self-limited disease. In an earlier report, a 14-month-old child infected by coxsackievirus B who was ultimately diagnosed as myocarditis presented with a tonic-clonic seizure as the prominent symptom [31]. In addition, the seizure occurred without fever, so it was identified not a febrile seizure. The cause of seizure in the patient may be related to transient hypoxia that resulted from supraventricular arrhythmia or heart block or cardiogenic embolism to the brain [32, 33]. However, the direct evidence of brain invasion by coxsackievirus was not found. However, this report suggested that seizure could be an initial presentation of myocarditis, which is important for the early diagnosis and treatment of myocarditis.
The infection with hepatitis A virus is a self-limited disease. However, in a 5-year-old patient infected with hepatitis A virus who complained the convulsions, RNA of hepatitis A virus was detected in the cerebrospinal fluid, which supported that the seizure activity resulted from hepatitis A virus infection [34] via toxic metabolic product accumulation and direct effect of the virus.
TMEV-infected acute seizures or epilepsy
Description of the TMEV-induced model
TMEV is a member of the Cardiovirus genus in the Picornaviridae family. In C57BL/6(B6) mice, TMEV infection is considered not to cause demyelinating disease, and the virus will be cleared in the early stage of acute encephalitis [35], while the TMEV-infected SJL mice are suspectable to demyelination and are used as a model of multiple sclerosis [36]. The strains of TMEV can be divided into two subgroups based on their neurovirulence after intracerebral inoculation: the less neurovirulent Theiler’s original (TO) group including the TO, Daniels (DA), BeAn 8386 (BeAn) and WW strains, and the more neurovirulent GDVII group including the GDVII and FA strains, which can induce fatal polioencephalomyelitis in short time in adult mice. In addition, a recent study characterized the geno- and phenotype of two naturally occurring variants of BeAn 8386 (BeAn-1 and BeAn-2) in the SJL/J and B6 mice, and found that the BeAn-1 substrain was more neurovirulent in SJL/J mice while BeAn-2 was more virulent in C57BL/6 mice [37]. These findings highlight that relatively minor variations in nucleotide sequence can lead to marked differences in neurovirulence.
A previous study showed that almost 51% of the C57BL/6 mice had transient afebrile seizures within 3 to 7 days [17]. Besides, TMEV infection also induced chronic seizures and altered seizure susceptibility [38]. The mice that develop seizures survived from the acute encephalitis and cleared the virus by 14 days after the infection with TMEV [39]. As to the frequency, the acute seizure lasted for 1 to 2 min during 2-h observation [17]. In addition, the percentage of C57BL/6 mice that developed seizures with DA strain infection is associated with the initial viral dose [39]. The TMEV-infected animals are likely to present with anxiety behaviors, poor performance in novel object exploration and impairment of episodic and spatial memory. Those psychoses have also been found in patients with epilepsy at a higher rate than the normal subjects [40]. Long-term video-EEG shows that a large proportion of TMEV-infected mice with epileptic episodes have experienced a distinct latent period in which no obvious seizures occur before the development of severe spontaneous epileptic seizures at 2, 4, and 7 months of age [41]. EEG recoding further revealed that the frequency of EEG spikes and spike clusters are higher in the DA-infected C57BL/6 mice with chronic seizures than in the infected mice without seizures [42]. Moreover, the TMEV-induced mice have dramatic cell death and gliosis in CA1 and CA2 pyramidal regions, but not in CA3 [17, 38], which is similar with the hippocampal sclerosis in patients with infection-induced temporal lobe epilepsy (TLE) [41]. These two prominent features are critical for the development of epilepsy and are considered to be a potential target for screening of patients for treatment [43]. Specifically, the gliosis includes astrogliosis and microgliosis [44].
The immune component involved in TMEV-induced seizures
Monocytes and granulocytes
In early studies, researchers noted that monocytes contributed to the development of epilepsy [35]. Peripheral circulating macrophages can infiltrate into the brains of the TMEV-infected mice at the onset of epilepsy, which is critical for epileptogenesis [45]. The similar situation has been observed in patients. The infiltration of monocytes has been found in the hippocampus of mesial TLE patients based on the biomarker CD45 [45]. The activated monocytes destroy the BBB, which increase the permeability of vessels to trigger the death of neurons and the generation of seizures [46]. A study monitored the kinetics of leukocyte infiltration during the first 48 h after TMEV infection, and found that the inflammatory monocytes appeared earlier than neutrophils [47]. Depletion of inflammatory monocytes and neutrophils resulted in the preservation of the hippocampus and maintenance of cognitive performance while depletion of neutrophils alone did not make any difference [48]. The administration of clodronate liposome that causes partly depletion of infiltrating macrophages by apoptosis in peripheral blood and the spleen decreases the rate of acute seizures in the TMEV-infected mice. But hippocampal damage seems not to be prevented. At the same time, the quantity of activated microglia in specific brain regions such as the hippocampus is found to increase as a response to the inhibition of macrophage infiltration. Indeed, microglia may have a protective function as they inhibit the development of seizures [49]. The different roles of infiltrating macrophages and microglia (resident cells) need to be explored deeply. The intrinsic relationship between infiltrating monocytes, neutrophils and macrophage is a critical element in the brain parenchyma injury. All of these cells are underlying points for the neuron protection and other comorbidities.
Therefore, the TMEV-induced mice are considered as an animal model that presents with many of the characteristics of encephalitis-induced human TLE, which can be used to investigate the relationship between inflammation and epilepsy. An obvious advantage of this model is that animals survive from the acute epilepsy, so the following research on chronic seizure can be carried out. In addition, neuronal degeneration in the CA1 pyramidal layer can be observed at 4 days post infection (dpi), while neurons were almost completely lost in the layer at 14 dpi [17, 41]. In another research, caspase-associated apoptosis has been found in CA1 neurons at 1 to 2 dpi, which may result from the oxidative stress, and this event has proceeded to the end stage of apoptotic cascade by 4 dpi. However, the apoptosis may not be mediated in a cell-autonomous manner, but be pathologically caused by the disruption of hippocampal circuits [50]. Seizures are results from the decreased inhibitory neural CA3 networks during the acute period [51]. However, increased excitation occurs both during the early infection and at 2 months later, when spontaneous epilepsy occurs in the exciting CA3 circuits [52]. The two different mechanisms suggest application of diverse treatments in the virus-infected patients.
Cytokines
Oxidative and nitrosative damage caused by decreased GSH and an increase in 3NT level are pivotal factors that lead to cell death in the hippocampus. The increase in 3NT levels indicates that the reactive oxygen species/reactive nitrogen species-induced post-translational modification may promote the occurrance of epilepsy [53]. In fact, the alteration of oxidative status is influenced by the direct viral damage. In addition, the increased levels of cytokines, chemokines, complements and other inflammatory mediators can also cause damage to mitochondria, resulting in oxidative stress, especially IL-6 and TNF-α.
The mRNA and protein levels of TNF-α are increased during acute epilepsy in the TMEV-infected animal model. The protein expression ratio of TNF receptors (TNFR) (TNFR1:TNFR2) is augmented as well. TNF-α can exert completely opposite functions through different receptors, including TNFR1 and TNFR2. TNF-α binding to TNFR1 can activate caspase-3, inducing apoptosis. TNFR2 mediates cell protection from apoptosis, resulting in the expression of inflammatory mediators and anti-apoptotic proteins. The two receptors also have cross talks and cooperate to transmit signals [54]. TNF-α may strengthen excitatory synapses via TNFR1 to increase the excitability in the hippocampus, while TNFR2 may provide adverse effects during acute infection [55]. In addition, mice without TNFR1 show inhibited seizures in this model, consistent with that in the chemical-induced seizure model (kainic acid seizure model) [45, 56].
It has been reported that the TMEV-infected mice lacking IL-6 may have reduced seizures, different from that in the kainic acid-induced mice [57]. This discrepancy may be explained by the different agents used to induce seizure, the different roles inflammatory mediators play, and the different genetic backgrounds of mice. IL-6 functions in the development of acute epilepsy to decrease group-II metabotropic glutamate receptors (mGluR2/3) and the AMPA receptor subunit 2 (GluA2) to increase glutamate release into the synapse, leading to strengthening of neuronal excitability [18]. Both resident CNS cells and infiltrating cells, which are recruited by chemokines, are important for the production of IL-6 in the brain and the development of seizures. Furthermore, macrophages are more likely to produce IL-6 standing for the infiltrating cells, while microglia and/or astrocytes are more likely to produce IL-6 standing for the resident CNS cells [58]. The main factors IL-6 and TNF-α are produced by both macrophages and microglia [59]. In particular, IL-6 mainly comes from infiltrating macrophages and microglia release TNF-α, inducing the onset of inflammation and then seizures. As mentioned above, the increased microglia can inhibit the macrophage infiltration, while the number of infiltrating macrophages does not decline.
Lymphocytes
The attack of T cells is also an important component of the immune system for viral clearance. However, CD8+ T cells are not the driving force to stop epilepsy by killing TMEV in OT-I mice (ovalbumin-specific CD8+ T cells) [45]. Above all, it is the innate immune response, rather than the adaptive immune response, that functions in the reduction of seizures.
Complement
The complement system, especially component 3 (C3), plays an important role in innate immune system. In addition, C3 can be released by many cells in the CNS, for example, astrocytes, microglia, neurons, and oligodendrocytes [60, 61]. After viral infection in the CNS, the expression of complement proteins is elevated [62]. Some researchers used C3-deficient C57BL/6 mice or depleted C3 in the periphery of C57BL/6 mic to evaluate whether C3 accelerates the process of seizure generation. No significance difference has been found between the C3-depleted mice and normal mice. The findings indicate that the activation promotes the development of seizures, which is not influenced by C3 in the periphery. The contribution of complement may be through the release of IL-6 and TNF-α [63].
Anti-inflammation treatment may slow down the process of epilepsy, or in other words, be disease-modifying. In contrast, drugs with direct antiseizure activity may reduce the presence of symptomatic seizures, without altering the disease at the initiation time [64].
As the drug resistance issue in the treatment of patients with epilepsy has been receiving more and more attention, a stable infection model such as the TMEV model is essential for the evaluation and modification of antiseizure agents. Many questions concerning chronic spontaneous seizures, particularly the molecular mechanisms, remain unanswered.
Treatment assessment
Minocycline (MIN) and valproic acid (VPA) affect epilepsy through different nodes in anti-inflammation. MIN treatment reduces microglial activation, which alleviates the long-term epileptogenic process [65]. MIN also decreases the number of infiltrating macrophages by approximately 2-fold. TNF-α, which is mostly produced by microglia and IL-6, which mainly comes from infiltrating macrophages, are also decreased by administration of MIN. The prolonged treatment with VAP has a neuroprotection effect through inhibition of histone deacetylase (HDAC) activity, leading to the low expression of GAPDH. Thus, the nuclear translocation of GAPDH that is related to cell apoptosis is decreased [66]. VAP at therapeutic doses can mitigate the acute seizure burden [67]. However, low-dose VAP did not show an obvious influence on the chronic epilepsy after TMEV infection, suggesting that VAP does not participate in the development of seizures. However, the MIN-treated mice showed improvement in long-term behavior and decreased seizure threshold [64].
Carbamazepine (CBZ) is one of the traditional anti-seizure drugs. It is intriguing that the number of CBZ-treated mice with seizure presentation increased compared with the vehicle-treated mice. In another word, CBZ exacerbated the infection of TMEV, making animals more susceptible to the virus [67]. CBZ has been found to contribute to the immunological defects and induce lymphopenia [68]. CBZ has also been shown to provoke epilepsy after Herpes simplex encephalitis in clinical studies [69]. As to the long-term comorbidities, administration of CBZ during the acute infection period also lead to development of behavioral impairments, such as worse anxiety behavior and spatial memory deficits. Not all traditional antiseizure drugs can be used for infection-induced seizures. These studies indicate that anti-inflammatory agents may be a preventive strategy for future epilepsy. The TMEV-infected animal model provides a platform to study the mechanism of epileptogenesis and to evaluate the disease-modifying functions of compounds.
Wogonin isolated from Scutellaria baicalensis has been found as an antiepileptic reagent. Wogonin functions through enhancement of GABA binding with the γ subunit [70], and has less adverse effects than diazepam such as sedative and muscle relaxant effects [71]. In the TMEV-infected animal model, wogonin exerts an anti-inflammatory effect like MIN. Wogonin can decrease the number of mice with epileptic episodes by inhibiting the activated macrophages targeted by the IL-6-producing cells [67]. The relationship between inflammation and GABA or other neurotransmitters likes norepinephrine, dopamine, and serotonin remains to be investigated.
Not only infiltrating macrophages are important components in the development of seizures following TMEV infection, but inflammatory cytokines are also critical factors in TMEV-induced seizures. Increased metabotropic glutamate receptor 5 in the hippocampus has been found in resected brain tissues from TLE patients, consistent with that in the TMEV-induced mice from 3 dpi to 14 dpi [72, 73]. Administration of VU0360172, a selective positive allosteric modulator, can reduce the acute seizures 3 days post infection through the TNF-α pathway, suggesting that VU0360172 have a neuroprotective function. However, the long-term treatment (8 dpi) did not induce a significant reduction [73].
Conclusions
Seizure represents a serious burden for individuals, families and the society. The acquired seizures, especially the encephalitis-associated seizures, should be prevented, ameliorated, and inhibited in the early stage. But the mechanisms of epileptogenesis are illusive. Unfortunately, there are many patients resistant to antiepileptic drugs. The traditional drugs are facing a rigorous challenge, and discovery of more targets is needed.
TMEV, which has been successfully used to establish a TLE model in the C57BL/6 mice, belongs to the Picornaviridae family. This model can be used as a tool to investigate the detailed components that may influence the process of seizure generation and to evaluate antiepileptic drugs. Some viruses of the Picornaviridae family can cause seizures as a prominent or atypical symptom, which should receive more attention.
The induction of seizures by virus infection in both mice and humans is directly caused by the excessive inflammation, which could lead to irreversible tissue damage, forming a big strike for limited regeneration of organs. Therefore, therapeutic strategies should focus on the inhibition of immune response and the innate response, which are critical for the development of seizures.
Availability of data and materials
Not applicable.
Abbreviations
- 3NT:
-
3-nitrotyrosine
- AMPA:
-
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- BBB:
-
Blood-brain barrier
- CBZ:
-
Carbamazepine
- CNS:
-
Central neurological system
- DA:
-
Daniels
- dpi:
-
Days post infection
- EEG:
-
Electroencephalogram
- GABA:
-
γ-aminobutyric acid
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase
- GluA2:
-
Glutamate AMPA receptor subunit 2
- GSH:
-
Glutathione
- HDAC:
-
Histone deacetylase
- HPeVs:
-
Human parechoviruses
- HRV:
-
Human rhinovirus
- IL-6:
-
Interleukin-6
- mGluR:
-
Metabotropic glutamate receptors
- MIN:
-
Minocycline
- OT-I:
-
Ovalbumin-specific CD8+ T cells
- TLE:
-
Temporal lobe epilepsy
- TMEV:
-
Theiler’s murine encephalomyelitis virus
- TNF-α:
-
Tumor necrosis factor-α
- TNFR:
-
TNF receptors
- TO:
-
Theiler’s original
- VPA:
-
Valproic acid
References
Wiebe S. Epilepsy: a comprehensive textbook on CD-ROM. Br Med J. 2000;320(7237):810.
Collaborators GBDE. Global, regional, and national burden of epilepsy, 1990-2016: a systematic analysis for the global burden of disease study 2016. Lancet Neurol. 2019;18(4):357–75.
Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58(4):512–21.
Bonello M, Michael BD, Solomon T. Infective causes of epilepsy. Semin Neurol. 2015;35(3):235–44.
Singhi P. Infectious causes of seizures and epilepsy in the developing world. Dev Med Child Neurol. 2011;53(7):600–9.
Misra UK, Tan CT, Kalita J. Viral encephalitis and epilepsy. Epilepsia. 2008;49(Suppl 6):13–8.
Lowenstein DH, Walker M, Waterhouse E. Status epilepticus in the setting of acute encephalitis. Epilepsy Curr. 2014;14(1 Suppl):43–9.
Tan CT, Goh KJ, Wong KT, Sarji SA, Chua KB, Chew NK, et al. Relapsed and late-onset Nipah encephalitis. Ann Neurol. 2002;51(6):703–8.
Chen SF, Huang CC, Wu HM, Chen SH, Liang YC, Hsu KS. Seizure, neuron loss, and mossy fiber sprouting in herpes simplex virus type 1-infected organotypic hippocampal cultures. Epilepsia. 2004;45(4):322–32.
Kalita J, Misra UK, Pandey S, Dhole TN. A comparison of clinical and radiological findings in adults and children with Japanese encephalitis. Arch Neurol. 2003;60(12):1760–4.
Pandey S, Rathore C, Michael BD. Antiepileptic drugs for the primary and secondary prevention of seizures in viral encephalitis. Cochrane Database Syst Rev. 2014;10:CD010247.
Getts DR, Balcar VJ, Matsumoto I, Mueller M, King NJC. Viruses and the immune system: their roles in seizure cascade development. J Neurochem. 2008;104(5):1167–76.
Adams MJ, Lefkowitz EJ, King AM, Harrach B, Harrison RL, Knowles NJ, et al. Ratification vote on taxonomic proposals to the international committee on taxonomy of viruses (2016). Arch Virol. 2016;161(10):2921–49.
Anastasina M, Domanska A, Palm K, Butcher S. Human picornaviruses associated with neurological diseases and their neutralization by antibodies. J Gen Virol. 2017;98(6):1145–58.
Victoria JG, Kapoor A, Li L, Blinkova O, Slikas B, Wang C, et al. Metagenomic analyses of viruses in stool samples from children with acute flaccid paralysis. J Virol. 2009;83(9):4642–51.
Gerhauser I, Hansmann F. Facets of Theiler's murine encephalomyelitis virus-induced diseases: an update. Int J Mol Sci. 2019;20(2):448.
Libbey JE, Kirkman NJ, Smith MC, Tanaka T, Wilcox KS, White HS, et al. Seizures following picornavirus infection. Epilepsia. 2008;49(6):1066–74.
DePaula-Silva AB, Hanak TJ, Libbey JE, Fujinami RS. Theiler's murine encephalomyelitis virus infection of SJL/J and C57BL/6J mice: models for multiple sclerosis and epilepsy. J Neuroimmunol. 2017;308:30–42.
Chew SP, Chong SL, Barbier S, Matthew A, Lee JH, Chan YH. Risk factors for severe hand foot mouth disease in Singapore: a case control study. J Neuro-Oncol. 2015;15:486.
Francis JR, Richmond P, Robins C, Lindsay K, Levy A, Effler PV, et al. An observational study of febrile seizures: the importance of viral infection and immunization. BMC Pediatr. 2016;16(1):202.
Britton PN, Jones CA, Macartney K, Cheng AC. Parechovirus: an important emerging infection in young infants. Med J Aust. 2018;208(8):365–9.
Hosoya M, Sato M, Honzumi K, Katayose M, Kawasaki Y, Sakuma H, et al. Association of nonpolio enteroviral infection in the central nervous system of children with febrile seizures. Pediatrics. 2001;107(1):E12.
Chen CS, Yao YC, Lin SC, Lee YP, Wang YF, Wang JR, et al. Retrograde axonal transport: a major transmission route of enterovirus 71 in mice. J Virol. 2007;81(17):8996–9003.
Chai Q, He WQ, Zhou M, Lu H, Fu ZF. Enhancement of blood-brain barrier permeability and reduction of tight junction protein expression are modulated by chemokines/cytokines induced by rabies virus infection. J Virol. 2014;88(9):4698–710.
Wahid R, Cannon MJ, Chow M. Dendritic cells and macrophages are productively infected by poliovirus. J Virol. 2005;79(1):401–9.
To KKW, Lau SKP, Chan K-H, Mok K-Y, Luk HKH, Yip CCY, et al. Pulmonary and extrapulmonary complications of human rhinovirus infection in critically ill patients. J Clin Virol. 2016;77:85–91.
Saghazadeh A, Gharedaghi M, Meysamie A, Bauer S, Rezaei N. Proinflammatory and anti-inflammatory cytokines in febrile seizures and epilepsy: systematic review and meta-analysis. Rev Neurosci. 2014;25(2):281–305.
Harvala H, Simmonds R. Human parechoviruses: biology, epidemiology and clinical significance. J Clin Virol. 2009;45(1):1–9.
Oh KW, Moon CH, Lee KY. Association of Rotavirus with seizures accompanied by cerebral White matter injury in neonates. J Child Neurol. 2015;30(11):1433–9.
Pariani E, Pellegrinelli L, Pugni L, Bini P, Perniciaro S, Bubba L, et al. Two cases of neonatal human PARECHOVIRUS 3 encephalitis. Pediatr Infect Dis J. 2014;33(11):1191–3.
Cunningham R, Silbergleit R. Viral myocarditis presenting with seizure and electrocardiographic findings of acute myocardial infarction in a 14-month-old child. Ann Emerg Med. 2000;35(6):618–22.
Shah SS, Hellenbrand WE, Gallagher PG. Atrial flutter complicating neonatal coxsackie B2 myocarditis. Pediatr Cardiol. 1998;19(2):185–6.
Butt AA, Solsi AC, Khan MA, Dukkipati MR, Dominguez A, Lazar EJ. Complete heart block and cardiogenic shock with coxsackievirus B4 myocarditis requiring permanent pacing and intra-aortic balloon counterpulsation. Am J Crit Care. 1995;4(4):319–21.
Cam S, Ertem D, Koroglu OA, Pehlivanoglu E. Hepatitis a virus infection presenting with seizures. Pediatr Infect Dis J. 2005;24(7):652–3.
Libbey JE, Fujinami RS. Neurotropic viral infections leading to epilepsy: focus on Theiler's murine encephalomyelitis virus. Futur Virol. 2011;6(11):1339–50.
Drescher KM, Sosnowska D. Being a mouse in a man's world: what TMEV has taught us about human disease. Front Biosci-Landmark. 2008;13:3775–85.
Broeer S, Hage E, Kaeufer C, Gerhauser I, Anjum M, Li L, et al. Viral mouse models of multiple sclerosis and epilepsy: marked differences in neuropathogenesis following infection with two naturally occurring variants of Theiler's virus BeAn strain. Neurobiol Dis. 2017;99:121–32.
Stewart K-AA, Wilcox KS, Fujinami RS, White HS. Theiler's virus infection chronically alters seizure susceptibility. Epilepsia. 2010;51(8):1418–28.
Libbey JE, Kennett NJ, Wilcox KS, White HS, Fujinami RS. Lack of correlation of central nervous system inflammation and neuropathology with the development of seizures following acute virus infection. J Virol. 2011;85(16):8149–57.
Umpierre AD, Remigio GJ, Dahle EJ, Bradford K, Alex AB, Smith MD, et al. Impaired cognitive ability and anxiety-like behavior following acute seizures in the Theiler's virus model of temporal lobe epilepsy. Neurobiol Dis. 2014;64:98–106.
Stewart K-AA, Wilcox KS, Fujinami RS, White HS. Development of Postinfection epilepsy after Theiler's virus infection of C57BL/6 mice. J Neuropathol Exp Neurol. 2010;69(12):1210–9.
Anjum SMM, Kaeufer C, Hopfengaertner R, Waltl I, Broeer S, Loescher W. Automated quantification of EEG spikes and spike clusters as a new read out in Theiler's virus mouse model of encephalitis-induced epilepsy. Epilepsy Behav. 2018;88:189–204.
Devinsky O, Vezzani A, Najjar S, De Lanerolle NC, Rogawski MA. Glia and epilepsy: excitability and inflammation. Trends Neurosci. 2013;36(3):174–84.
Loewen JL, Barker-Haliski ML, Dahle EJ, White HS, Wilcox KS. Neuronal injury, gliosis, and glial proliferation in two models of temporal lobe epilepsy. J Neuropathol Exp Neurol. 2016;75(4):366–78.
Cusick MF, Libbey JE, Patel DC, Doty DJ, Fujinami RS. Infiltrating macrophages are key to the development of seizures following virus infection. J Virol. 2013;87(3):1849–60.
Marchi N, Angelov L, Masaryk T, Fazio V, Granata T, Hernandez N, et al. Seizure-promoting effect of blood-brain barrier disruption. Epilepsia. 2007;48(4):732–42.
Howe CL, LaFrance-Corey RG, Sundsbak RS, Sauer BM, LaFrance SJ, Buenz EJ, et al. Hippocampal protection in mice with an attenuated inflammatory monocyte response to acute CNS picornavirus infection. Sci Rep. 2012;2(1).
Howe CL, LaFrance-Corey RG, Sundsbak RS, LaFrance SJ. Inflammatory monocytes damage the hippocampus during acute picornavirus infection of the brain. J Neuroinflammation. 2012;9(1).
Waltl I, Kaeufer C, Gerhauser I, Chhatbar C, Ghita L, Kalinke U, et al. Microglia have a protective role in viral encephalitis-induced seizure development and hippocampal damage. Brain Behav Immun. 2018;74:186–204.
Buenz EJ, Sauer BM, LaFrance-Corey RG, Deb C, Denic A, German CL, et al. Apoptosis of hippocampal pyramidal neurons is virus independent in a mouse model of acute Neurovirulent Picornavirus infection. Am J Pathol. 2009;175(2):668–84.
Smeal RM, Fujinami R, White HS, Wilcox KS. Decrease in CA3 inhibitory network activity during Theiler's virus encephalitis. Neurosci Lett. 2015;609:210–5.
Smeal RM, Stewart K-A, Iacob E, Fujinami RS, White HS, Wilcox KS. The activity within the CA3 excitatory network during Theiler's virus encephalitis is distinct from that observed during chronic epilepsy. J Neuro-Oncol. 2012;18(1):30–44.
Bhuyan P, Patel DC, Wilcox KS, Patel M. Oxidative stress in murine Theiler's virus-induced temporal lobe epilepsy. Exp Neurol. 2015;271:329–34.
Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol. 2003;3(9):745–56.
Patel DC, Wallis G, Dahle EJ, McElroy PB, Thomson KE, Tesi RJ, et al. Hippocampal TNF alpha signaling contributes to seizure generation in an infection-induced mouse model of limbic epilepsy. Eneuro. 2017;4(2):ENEURO.0105-17.2017. eCollection Mar-Apr 2017.
Balosso S, Ravizza T, Perego C, Peschon J, Campbell IL, De Simoni MG, et al. Tumor necrosis factor-alpha inhibits seizures in mice via p75 receptors. Ann Neurol. 2005;57(6):804–12.
Penkowa M, Molinero A, Carrasco J, Hidalgo J. Interleukin-6 deficiency reduces the brain inflammatory response and increases oxidative stress and neurodegeneration after kainic acid-induced seizures. Neuroscience. 2001;102(4):805–18.
Libbey JE, Kennett NJ, Wilcox KS, White HS, Fujinami RS. Interleukin-6, produced by resident cells of the central nervous system and infiltrating cells, contributes to the development of seizures following viral infection. J Virol. 2011;85(14):6913–22.
Zattoni M, Mura ML, Deprez F, Schwendener RA, Engelhardt B, Frei K, et al. Brain infiltration of leukocytes contributes to the pathophysiology of temporal lobe epilepsy. J Neurosci. 2011;31(11):4037–50.
Yu JX, Bradt BM, Cooper NR. Constitutive expression of proinflammatory complement components by subsets of neurons in the central nervous system. J Neuroimmunol. 2002;123(1–2):91–101.
Hosokawa M, Klegeris A, Maguire J, McGeer PL. Expression of complement messenger RNAs and proteins by human oligodendroglial cells. Glia. 2003;42(4):417–23.
Dietzschold B, Schwaeble W, Schafer MK, Hooper DC, Zehng YM, Petry F, et al. Expression of C1q, a subcomponent of the rat complement system, is dramatically enhanced in brains of rats with either Borna disease or experimental allergic encephalomyelitis. J Neurol Sci. 1995;130(1):11–6.
Libbey JE, Kirkman NJ, Wilcox KS, White HS, Fujinami RS. Role for complement in the development of seizures following acute viral infection. J Virol. 2010;84(13):6452–60.
Barker-Haliski ML, Heck TD, Dahle EJ, Vanegas F, Pruess TH, Wilcox KS, et al. Acute treatment with minocycline, but not valproic acid, improves long-term behavioral outcomes in the Theiler's virus model of temporal lobe epilepsy. Epilepsia. 2016;57(12):1958–67.
Wang N, Mi X, Gao B, Gu J, Wang W, Zhang Y, et al. Minocycline inhibits brain inflammation and attenuates spontaneous recurrent seizures following pilocarpine-induced status epilepticus. Neuroscience. 2015;287:144–56.
Kanai H, Sawa A, Chen RW, Leeds P, Chuang DM. Valproic acid inhibits histone deacetylase activity and suppresses excitotoxicity-induced GAPDH nuclear accumulation and apoptotic death in neurons. Pharmacog J. 2004;4(5):336–44.
Barker-Haliski ML, Dahle EJ, Heck TD, Pruess TH, Vanegas F, Wilcox KS, et al. Evaluating an etiologically relevant platform for therapy development for temporal lobe epilepsy: effects of carbamazepine and Valproic acid on acute seizures and chronic behavioral comorbidities in the Theiler's murine encephalomyelitis virus mouse model. J Pharmacol Exp Ther. 2015;353(2):318–29.
Spickett GP, Gompels MM, Saunders PWG. Hypogammaglobulinaemia with absent B lymphocytes and agranulocytosis after carbamazepine treatment. J Neurol Neurosurg Psychiatry. 1996;60(4):459.
Rice CM, Johnston SL, Unsworth DJ, Glover SC, Donati M, Renowden SA, et al. Recurrent herpes simplex virus encephalitis secondary to carbamazepine induced hypogammaglobulinaemia. J Neurol Neurosurg Psychiatry. 2007;78(9):1011–2.
Tai MC, Tsang SY, Chang LYF, Xue H. Therapeutic potential of wogonin: a naturally occurring flavonoid. Cns Drug Rev. 2005;11(2):141–50.
Cheong JH, Ryu JH, Ko KH, Ko HS, Yoon SY, Lim BW, et al. Different effects of flavonoids in Scutellaria baicalensis on anxious and sedative behaviors. Biomol Ther. 2006;14(2):75–81.
Kandratavicius L, Rosa-Neto P, Monteiro MR, Guiot M-C, Assirati JA Jr, Carlotti CG Jr, et al. Distinct increased metabotropic glutamate receptor type 5 (mGluR5) in temporal lobe epilepsy with and without hippocampal sclerosis. Hippocampus. 2013;23(12):1212–30.
Hanak TJ, Libbey JE, Doty DJ, Sim JT, DePaula-Silva AB, Fujinami RS. Positive modulation of mGluR5 attenuates seizures and reduces TNF-alpha(+) macrophages and microglia in the brain in a murine model of virus-induced temporal lobe epilepsy. Exp Neurol. 2019;311:194–204.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural Science Foundation of Shandong, China (ZR2019PH040) and the National Natural Science Foundation of China (81901321).
Author information
Authors and Affiliations
Contributions
XSC designed the review, and revised the manuscript. RXZ and JC conducted the systematic search and extracted the eligible studies. RXZ drafted the study. JM and WJJ revised the manuscript. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors gave consent to publication of this review.
Competing interests
The authors declare no conflicts of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, R., Mu, J., Chi, J. et al. The role of picornavirus infection in epileptogenesis. Acta Epileptologica 3, 6 (2021). https://doi.org/10.1186/s42494-021-00040-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42494-021-00040-6