
Abstract
Yam mild mosaic virus (YMMV) is prevalent in yams (Dioscorea spp.) worldwide. To gain an insight into the genetic diversity and molecular evolution of YMMV, 89 isolates from West Africa, Asia, South Pacific, and America were analyzed phylogenetically by using sequence between the 3′-terminal of the coding region of the coat protein and the 5′-terminal of non-coding region at the 3′ end of the YMMV genome. The results revealed that there was a significant genetic diversity among isolates and a clear correlation between the coat protein gene sequence and the geographical origin of YMMV isolates. Of particular, YMMV isolates from North China and South China fell into two different groups. Furthermore, full genome comparison identified four chimeric genome patterns and six putative recombination signals representing four recombination events were detected among 12 genomes of independent isolates from China and Brazil, suggesting a high frequency of genome recombination event.
Similar content being viewed by others


Background
Yam mild mosaic virus (YMMV), a distinct member of the genus Potyvirus, is a major viral agent in yams in Africa, Asia, Oceania, Caribbean and South America (Mumford and Seal 1997; Fuji et al. 1999; Odu et al. 1999; Bousalem and Dallot 2000; Dallot et al. 2001; Eni et al. 2008; Zou et al. 2011). This virus has flexuous, filamentous particles of approximately 750 nm in length and is transmitted by aphids or mechanical inoculation (Odu et al. 1999). The virus causes mild symptoms of mottle and mosaic on water yam (Dioscorea alata), Guinea yams (D. cayenensis – D. rotundata complex) and Indian yam (D. trifida), but no symptoms on white yam (D. rotundata) (Mumford and Seal 1997). Its natural host range is restricted to several Dioscorea spp., but the virus is also transmitted easily to cowpea (Vigna unguiculata) (Odu et al. 1999). It was reported that YMMV isolates from Caribbean island Martinique and French Guyana were divergent in their coat protein (CP) gene sequences (Bousalem et al. 2003). However, little was known about the diversity of the YMMV at whole genome level, simply because only three whole genome sequences of YMMV were available at the time (Simon-Loriere and Holmes 2011; Filho et al. 2013).
The Qinling Mountains-Huaihe River Line, with its west end at E104°15′/N32°18′ and east end at E120°21′/N34°05′separates China as South (south to the line) and North (north to the line) mainly by its climatic impact. Yams are mainly grown in the provinces of Guangxi and Jiangxi in South China, and Henan, Shandong and Jiangsu in North China. YMMV infection of yam plants in China was first reported in 2010 (Zou et al. 2011). To gain an insight into the composition of genetic population of YMMV, we conducted a nation-wide survey of YMMV in China. Here, we report the identification, genome sequencing, phylogenetic analysis, and genome recombination analysis of YMMV isolates from China and those reported elsewhere. Our results showed that YMMV genome sequences were diversified among isolates with geographical characteristics, and extensive genome recombination events had taken place in the population of the virus.
Results
YMMV infection was prevalent in yams in the major yam-producing regions in China
During the survey, leaves and tubers of yam plants showing viral disease-like symptoms, i.e., mosaic, chlorosis, vein banding, flecking, leaf puckering, stunting and distortion were collected, with leaves stored at − 80 °C for viral identification and tubers grown in the glasshouse for keeping the materials. IC-RT-PCR and sequencing results revealed that 112 out of 365 samples collected from five yam producing regions (Guangxi, Jiangxi, Henan, Jiangsu and Shandong) in China were YMMV-positive, among which 75 were from 132 D. alata, 11 from 96 D. japonica and 26 from 137 D. opposite. The incidence ranged from 11% to 40% in general, varying among yam species with the highest rate for D. alata and lowest for D. japonica (Additional file 1: Table S1). Most YMMV-positive samples (90.5%) were co-infected with other viruses (our unpublished results) and those (9.5%) solely infected with YMMV showed constantly a mild mosaic or mottle or symptomless on leaves (Fig. 1), regardless of species/cultivars and years of sampling. The rest of YMMV-negative samples were infected either with other known viruses, uncharacterized viruses, or unknown agents.

Symptoms of yam leaves infected with YMMV. All plants were infected solely by YMMV. a Cultivar Guihuai 2 (D. japonica); b Cultivar Guihuai 6 (D. alata); c Cultivar Tiegun Shanyao (D. opposite)
YMMV isolates showed a clear geographical character
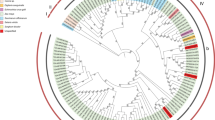
To establish evolutionary relationship among the YMMV isolates, sequences from the region (nucleotides 259–262) in the 3′-terminal region of CP to the 5′-terminal region of 3′-nocoding region (NCR) of 26 YMMV isolates obtained in this study and 37 isolates previously reported, and 26 unpublished YMMV isolates (Table 1) were used to construct an unrooted phylogenetic tree using Maximum Likelihood algorithm. Among 14 groups clustered in the phylogenetic tree (Fig. 2), YMMV isolates from China fall into two groups: Group VII that contains isolates from North China, and Group X that contains isolates from South China, both distinguishing themselves from isolates from West Africa (Group II, Group III, Group XI, Group XII, Group XIII, and Group XIV), America (Group IV and Group VI), and other parts of Asia (Group V and Group VIII).

Phylogenetic relationships of 89 isolates representing 14 YMMV groups. Maximum-likelihood phylogenetic tree based on the partial coat protein and 3′-UTR sequences (259 to 262 nucleotides) of 26 isolates from China and 65 isolates sequenced previously (Table 1, sequences were available from GenBank as of Jan, 2020). The sequence accession numbers for each of the isolates were shown in Table 1. Bootstrap values were for 1000 replicates
YMMV population was diversified at whole genome level
The genome sequences of isolates representing Group X and Group VII were assembled from the high throughput sequencing (HTS) data of the infected yam leaf samples and validated by RT-PCR and RACE. The whole genomes of the 12 YMMV isolates were from 9521 to 9538 nucleotides (nts) in length excluding the poly (A) tail (GenBank Accession No. KC407674, KC473517, KJ125472–125479, JX470965, KX156847). Eleven of the 12 isolates were the same as the reported Brazilian isolate in terms of processed protein sizes, but with slightly different length in untranslated regions (UTRs) and isolate CN20 differed from the rest of the isolates in that its P1 protein was composed of 321 aa, instead of 320 aa (Additional file 2: Table S2). While motifs of FRNK in the HC-Pro, GDD in the NIb, and DAG in the CP were conserved in all isolates, residues flanking the motif of the HC-Pro for WX1, WX3, and XZ1 (Group VII) differed from other isolates, and more diversified patterns flanking the CP were seen (Fig. 3).

Schematic representation of YMMV genome organization and conserved motifs among 12 aligned genomes. Potyvirus motifs of FRNK in the HC-Pro, GDD in the NIb, and DAG in the CP were conserved in all sequenced YMMV isolates
Phylogenetic tree based on the whole genome sequences of YMMV isolates further showed that isolates from South China and North China grouped separately, with the North China group closer to the Brazilian and the Korean isolates (Fig. 4). By taking advantage of whole genome sequences of multiple isolates in Group VII and Group X, intra-group identity percentage of functional gene/protein and UTR at nucleotide and amino acid levels were calculated and compared with the Brazilian strain, the representative of Group IV (Additional file 3: Table S3 and Additional file 4: Table S4). As summarized in Table 2, at the whole genome level, identities were 87.8–99.7% for isolates within Group VII and 86.1–97.6% for isolates within Group X. However, identity between Group VII and Group X was only 84.9–87.5%. Even lower levels of identity were found between Group X and Group IV (82.8–83.6%) or between Group VII and Group IV (83.9–84.4%). It was noticed that 5′-UTRs were the most divergent, 65.1–96.2% within groups and 58.0–82.0% between groups. As to the Brazilian strain (group IV), it shares up to 84.4% identity to either Group X or Group VII, lower than that between Group X and Group VII. Less variation was found at amino acid level for the viral proteins of the isolates, both within and among groups of isolates, with the highest identity of 93.7–99.0% for isolates from Group X and 94.6–99.9% for isolates from Group VII at polyprotein level. Among the mature proteins, 6 K1 was the most conserved with an identity of 96.1–100% for isolates in Group X and 98.0–100% for isolates in Group VII, while P1 was the most variable, 77.8–99.4% for isolates in Group VII. An identity of 71.2–79.1% for P1 between Group X and Group VII was the lowest among all processed proteins. Brazilian strain shares an identity up to 91.3–92.0% to Group X or Group VII at polyprotein level, lower than the 92.6–93.9% between Group X and Group VII.

Phylogenetic tree based on the complete genomic sequences of 12 YMMV isolates. Maximum-likelihood phylogenetic tree based on the available complete genomic sequences of ten YMMV isolates from China and one isolate from Brazil
Genome recombination events in YMMV
Among the 12 genomes of YMMV isolates (ten from China, one from Brazil and one from Korea), three isolates (NN1, FX1 and NC1) seemed to have a “pure” genome, i.e. without any apparent chimeric genome fragment from the known isolates (Fig. 5). Six clear recombination signals representing four recombination events involving four YMMV genomes were detected with P-value < 1.0 × 10− 6 (Table 3). CP was the hottest region for recombination, involving five isolates, and P3 and CI each occurred once in one of the isolates. As shown in Fig. 5, one recombination event was found in NC1, CN1, NC2 and CN20, respectively. And there were two recombination regions in CN1 and CN20, which were located at both ends of the genome, respectively. There was also a recombination event in NC1 and NC2, but only one recombination region existed in each recombination event. The region of recombination event I was located at the 5 ‘end of NC1 genome, while the region of recombination event III was located at the middle and rear of NC2 genome.

Recombination in full-length YMMV genome sequences. The graph at the top shows an YMMV genomic map. Locations of unique recombination events were identified by RDP4, in relation to the full-length sequence alignment of 12 YMMV isolates. Each full-length genome is represented by a long colored bar and the corresponding isolate name, given to the left of the bar. The figure shows a total of 4 unique recombination events, demarcated by the bars below the genomes that the recombinant fragments have been integrated into. When an ancestral unique recombination event can be found in more than one daughter sequence, the recombination event is displayed with all corresponding daughter sequences. Locations of the unique recombination events identified by RDP4, corresponding to the manually verified recombination sites, are shown with distinguished colored bars
Discussion
Geographical distribution of YMMV isolates and movement of yam germplasm
YMMV is a world-wide dispersed potyvirus in yams, but not much is known about its natural history and evolutionary relation. A powerful tool for tracing the origin and evolution of a virus is phylogenetic and phylodynamic analysis of viral sequences (Ren et al. 2013). Data obtained using such methods have contributed to the surveillance of viral spread and drug resistance as well as the identification of strains as vaccine candidates (Lam et al. 2010; Norström et al. 2012).
As the 3′-terminal region of CP and 5′-terminal region of 3′-NCR of YMMV has been used for genotype classification in YMMV to investigate the diversity of YMMV (Bousalem et al. 2003), these sequences were used in this study to establish evolutionary relationship of YMMV isolates. As shown in Fig. 2, the 14 distinct groups clustered were strongly associated with geographical distribution, among which Group X was from South China, Group VII from North China. Most of the isolates from other Asia regions were closer to Chinese isolates than to those from the Central America and West Africa, indicating that the differentiation of YMMV was resulted from geographic isolation. Of particular note is that Group VII isolates from North China represent the most distant relation to the Group X from South China, and these two groups are different from those outside China, suggesting that YMMV isolates from China may have undergone further differentiation. The phylogenetic tree constructed from this study was partially in accordance with the assumption that Asian-Pacific origin of YMMV was likely from D. alata species (Bousalem et al. 2003). But our data also showed that YMMV isolates from China and India share a common ancient ancestor, different from those YMMV isolates from Central America and West Africa which share the same common ancient ancestor. Distinct geographical distribution of YMMV groups also suggests that germplasm exchange of yams has been infrequent between South China and North China, as well as among countries world-wide.
Phylogenetic relation and viral recombination at whole-genome scale among YMMV isolates
To address the genetic relation among different genotypes further, we employed 12 YMMV complete genome sequences to analyze the phylogenetic relationship. The predicted sizes of the coding regions were identical among YMMV isolates except CN20 (Additional file 2: Table S2). The extent of genetic diversity, reflected in percentage identity, varies within and among proteins, in the order NIa-VPg > HC-Pro > NIa-Pro > 6 K1 > CI > NIb > CP > 6 K2 > PIPO > P3 > P1 (Table 2). The phylogenetic tree constructed based on the whole genomes (Fig. 3) matches basically well with the tree constructed based on the partial CP core region and the 3′-NCR sequences (Fig. 2), with isolates from South China (NN1, NC1, NC2, NC3, FX1 and CN1) in Group X, isolates from North China (CN20, XZ1, WX1 and WX3) in group VII, and Brazilian isolate in group IV. The only exception is the isolate CN20, the placing of which is inconsistent in the two trees, suggesting a mixed genome. YMMV genome recombination was first observed by comparison of the partial 3′-teminal genome sequences of the isolates collected from different geographical locations (Bousalem et al. 2003). Indeed, in the current study, a detailed examination revealed that 4 out of the 12 YMMV isolates may have gone through genome recombination events (Fig. 5). While recombination events were spotted in the genome regions encoding P3, CI and CP, the most frequent recombination events were found in the CP-encoding 3′-end region. Viral recombination is a powerful contributor to genetic variation, adaptation to new hosts, escape from the host immune response, and emergence of newly infectious agents (Becher et al. 2001; Simon-Loriere and Holmes 2011). Although recombinants did not seem to have a significant impact on symptoms (Fig. 1), recombination in CP between YMMV isolates may provide a selective advantage for virus dissemination by a vector to adapt to the local environment, although the molecular mechanisms of this hypothesis needs to be clarified.
Conclusions
Data presented in this study demonstrated that YMMV infection was prevalent in the main yam producing areas in China, and there was a significant genetic diversity and a clear correlation between the coat protein gene sequence and the geographical origin of YMMV isolates. Four chimeric genome patterns were identified in 12 isolates, suggesting a high frequency of genome recombination event. Therefore, due cautions should be taken in the exchange of germplasms between the north and the south of China, as well as among nations in the world, to prevent possible occurrence of new virulent isolates through genome recombination between and among YMMV isolate types.
Methods
Sample collection and virus identification
During a survey of viral diseases on yam in 2010–2015, a total of 365 yam leaf samples were collected from Guangxi Province, Henan Province, Jiangsu Province, Jiangxi Province and Shandong Province in China, and 105 samples were identified as YMMV positive by IC-RT-PCR using polyclonal antibodies developed in our laboratory (Zou et al. 2011).
Nucleic acid extraction and analysis
Total RNA was extracted from an amount of 100 mg of yam leaf tissue using an RNAprep pure Plant Kit (Tiangen Inc., Beijing, China) and quantified by agarose gel electrophoresis and Qubit® 2.0 Fluorometer quantitation assay. Viral cDNA was synthesized with the total RNA as template using Transcriptor High Fidelity cDNA Synthesis Kit (Roche Applied Science, Mannhelm, Germany) and PCR amplification of YMMV genome fragments was performed using AmpliTaq DNA polymerase and the Expand high-fidelity PCR system (Roche Applied Science, Mannhelm, Germany) with virus-specific primers (Additional file 5: Table S5).
Deep sequencing, RACE and assembly of viral genome from total yam RNA
An amount of 5 μg of the total RNA was used for cDNA library construction using TruSeq Illumina mRNA library construction kit (Illumina Inc., San Diego, CA). Deep sequencing was performed on an Illumina Solexa GAIIx platform. The CLC Genomics Workbench V6.0.1 software was used for deep sequencing data analysis. Raw reads from the Illumina RNA-Seq were trimmed to remove low quality reads and sequencing adaptor. The clean reads were assembled into contigs using the De novo Assembly algorithm. Contigs were then mapped to the YMMV reference sequence (Genbank accession No. JX470965).
RACE amplification was performed using the SMARTer™ RACE cDNA Amplification Kit (Clontech Laboratories, Inc.). PCRs were carried out on cDNA with Phusion® High-Fidelity PCR Master Mix with HF Buffer (Thermo Fisher Scientific, Inc.). PCR fragments were purified from agarose gels and cloned into pJET1.2/blunt Cloning Vector (Thermo Fisher Scientific, Inc.). Sequences from each isolate were confirmed by analysis of at least three overlapping independent RT-PCR products on an Applied Biosystems 3730XL DNA Sequencer. Overlapped sequences were assembled and analyzed with Vector NTI Advance™ version 10 software (Invitrogen Inc., Carlsbad, CA).
Alignment and phylogenetic analysis
Nucleotide sequences of YMMV isolates were identified by BLAST search against NCBI (http://blast.ncbi.nlm.nih.gov/Blast.cgi) and conserved domain database (CDD) were used for conserved domain identification and structure analysis. DNAstar (DNASTAR, USA) was used to analyze nucleotide and amino acid sequence divergence. Alignment of the nucleotide or amino acid sequences was performed using the Clustal W program (Thompson et al. 1994). Phylogenetic analysis was performed with MEGA6 software using the maximum likelihood method with bootstrap of 1000 times repeat (Tamura et al. 2011), using CP/3′-UTR region (Mumford and Seal 1997) or the whole genome of the virus.
Genome recombination analysis
A collection of isolate genomes was first aligned using Clustal W program, then scanned by Recombination Detection Program version 4 (RDP4) with default settings for the different detection methods and a Bonferroni corrected P value cut-off of 0.05. (Martin et al. 2015). And the detected site with a Bonferroni corrected P values of less than 1.0 × 10− 6 was considered a clear recombination site, otherwise it was considered as a tentative recombination sites (Ohshima et al. 2007).
Availability of data and materials
Not applicable.
Abbreviations
- CP:
-
Coat protein
- HTS:
-
High throughput sequencing
- IC-RT-PCR:
-
Immunocapture Reverse transcription polymerase chain reaction
- NCR:
-
Nocoding region
- RT-PCR:
-
Reverse transcription polymerase chain reaction
- YMMV:
-
Yam mild mosaic virus
References
Becher P, Orlich M, Thiel H-J. RNA recombination between persisting pestivirus and a vaccine strain: generation of cytopathogenic virus and induction of lethal disease. J Virol. 2001;75:6256–64.
Bousalem M, Dallot S. First report and molecular characterization of Yam mild mosaic virus in Dioscorea alata on the island of Martinique. Plant Dis. 2000;84:200.
Bousalem M, Dallot S, Fuji S, Natsuaki KT. Origin, world-wide dispersion, bio-geographical diversification, radiation and recombination: an evolutionary history of Yam mild mosaic virus (YMMV). Infect Genet Evol. 2003;3:189–206.
Dallot S, Guzmán M, Bousalem M. Occurrence of potyviruses on yam (Dioscorea spp.) in Colombia and first molecular characterization of Yam mild mosaic virus. Plant Dis. 2001;85:803.
Eni AO, Hughes J’A, Rey MEC. Survey of the incidence and distribution of five viruses infecting yams in the major yam-producing zones in Benin. Ann Appl Biol. 2008;153:223–32.
Filho FACR, Nicolini C, Resende RO, Andrade GP, Pio-Ribeiro G, Nagata T. The complete genome sequence of a Brazilian isolate of yam mild mosaic virus. Arch Virol. 2013;158:515–8.
Fuji S, Mitobe I, Nakamae H, Natsuaki KT. Nucleotide sequence of coat protein gene of yam mild mosaic virus, isolated in Papua New Guinea. Arch Virol. 1999;144:1415–9.
Lam TT-Y, Hon C-C, Tang JW. Use of phylogenetics in the molecular epidemiology and evolutionary studies of viral infections. Crit Rev Clin Lab Sci. 2010;47:5–49.
Martin DP, Murrell B, Golden M, Khoosal A, Muhire B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015;1:vev003.
Mumford RA, Seal SE. Rapid single-tube immunocapture RT-PCR for the detection of two yam potyviruses. J Virol Methods. 1997;69:73–9.
Norström MM, Karlsson AC, Salemi M. Towards a new paradigm linking virus molecular evolution and pathogenesis: experimental design and phylodynamic inference. New Microbiol. 2012;35:101–11.
Odu BO, Hughes J’A, Shoyinka SA, Dongo LN. Isolation, characterisation and identification of a potyvirus from Dioscorea alata L. (water yam) in Nigeria. Ann Appl Biol. 1999;134:65–71.
Ohshima K, Tomitaka Y, Wood JT, Minematsu Y, Kajiyama H, Tomimura K, et al. Patterns of recombination in turnip mosaic virus genomic sequences indicate hotspots of recombination. J Gen Virol. 2007;88:298–315.
Ren L, Xiao Y, Li J, Chen L, Zhang J, Vernet G, et al. Multiple genomic recombination events in the evolution of saffold cardiovirus. PLoS One. 2013;8:e74947.
Simon-Loriere E, Holmes EC. Why do RNA viruses recombine? Nat Rev Microbiol. 2011;9:617–26.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–9.
Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–80.
Wang M, Li F, Zhou G, Lan P, Xu D, Li R. Molecular detection and characterization of Chinese yam mild mosaic virus isolates:n/a-n/a. J Phytopathol. 2015;163:1036–40.
Zou C, Meng J, Li Z, Wei M, Song J, Chen B, et al. First report of Yam mild mosaic virus in yam in Guangxi Province. China Plant Dis. 2011;95:1320.
Acknowledgements
We thank Pu Li (Guangxi University) for assistance in the analysis of deep sequencing data; Yong-Mei Liao (Guangxi University), Xin-Min Shi (Xuzhou Institute of Agricultural Sciences), Xin-Lin Dai and Jie Tang (Crops Research Institute, Jiangxi Academy of Agricultural Sciences), Ruo-Rui Qiu and Ai-Xian Li (Crops Research Institute, Shandong Academy of Agriculture), Suo-Yi Han (Industrial Crops Research Institute, Henan Academy of Agricultural Sciences), Su-Xia Wang (Modern Agriculture Experimental Station of Wen County, Henan), and Jian-He Cai (Institute of Plant Protection, Guangxi Academy of Agriculture) for helping the collection of yam samples.
Funding
This work was supported in part by grants from the National Public Service (agricultural) Industry Research Special Project of China (200903022) and Natural Science Foundation of Guangxi Province (2016GXNSFAA380027).
Author information
Authors and Affiliations
Contributions
BSC conceived the study and revised the manuscript. CWZ and LZ performed the experiments. CWZ, JRM and ZTY drafted the manuscript. JRM, ZQW and BHW contributed reagents/materials/analysis tools. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Supplementary information
Additional file 1: Table S1.
Symptom-showing samples collected and YMMV infection rate detected by IC-RT-PCR.
Additional file 2: Table S2.
Genome features of YMMV isolates.
Additional file 3: Table S3.
Nucleotide identity within YMMV isolate groups.
Additional file 4: Table S4.
Amino acid identity within YMMV isolate groups.
Additional file 5. Table S5.
Primers used in this work.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zou, CW., Meng, JR., Yao, ZT. et al. Genetic diversity and genome recombination in Yam mild mosaic virus isolates. Phytopathol Res 2, 10 (2020). https://doi.org/10.1186/s42483-020-00051-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42483-020-00051-0