
Abstract
Hereditary transthyretin (TTR) amyloidosis (ATTRv) is an autosomal dominant, systemic disease transmitted by amyloidogenic mutations in the TTR gene. To prevent the otherwise fatal disease course, TTR stabilizers and mRNA silencing antisense drugs are currently approved treatment options. With 90% of the amyloidogenic protein produced by the liver, disease progression including polyneuropathy and cardiomyopathy, the two most prominent manifestations, can successfully be halted by hepatic drug targeting or—formerly—liver transplantation. Certain TTR variants, however, favor disease manifestations in the central nervous system (CNS) or eyes, which is mostly associated with TTR production in the choroid plexus and retina. These compartments cannot be sufficiently reached by any of the approved medications. From liver-transplanted patients, we have learned that with longer lifespans, such CNS manifestations become more relevant over time, even if the underlying TTR mutation is not primarily associated with such. Are we therefore creating a new phenotype? Prolonging life will most likely lead to a shift in the phenotypic spectrum, enabling manifestations like blindness, dementia, and cerebral hemorrhage to come out of the disease background. To overcome the first therapeutic limitation, the blood–brain barrier, we might be able to learn from other antisense drugs currently being used in research or even being approved for primary neurodegenerative CNS diseases like spinal muscular atrophy or Alzheimer’s disease. But what effects will unselective CNS TTR knock-down have considering its role in neuroprotection? A potential approach to overcome this second limitiation might be allele-specific targeting, which is, however, still far from clinical trials. Ethical standpoints underline the need for seamless data collection to enable more evidence-based decisions and for thoughtful consenting in research and clinical practice. We conclude that the current advances in treating ATTRv amyloidosis have become a meaningful example for mechanism-based treatment. With its great success in improving patient life spans, we will still have to face new challenges including shifts in the phenotype spectrum and the ongoing need for improved treatment precision. Further investigation is needed to address these closed barriers and open questions.
Similar content being viewed by others

Background
Transthyretin (TTR) amyloidosis is a systemic disease caused by dissociation of the TTR tetramer and subsequent fibril deposition in tissues [1]. Despite the existence of a less aggressive disease form associated with wild-type TTR [2], this mechanism is attributed to more than 130 known amyloidogenic missense mutations in the TTR gene (OMIM *176300). With an autosomal dominant mode of inheritance, hereditary TTR (ATTRv) amyloidosis typically becomes symptomatic with a progressive sensorimotor and autonomic neuropathy and cardiac dysfunction [1]. Rarer manifestations include proteinuria, vitreous opacities, cerebral hemorrhage, stroke-like episodes, and dementia [3, 4]. It is so far unknown, why ATTR amyloid preferentially deposits in certain organs but not in others. Manifestation subtypes can loosely be correlated with the respective underlying TTR variant; however, this is not the only factor of influence. While the genetic cause of ATTRv amyloidosis itself is well understood and even targetable by several treatment options, important aspects like differences in organ tropism and manifestation onset are not well understood to date. As potential modifiers, influencing factors in the extracellular matrix, in the TTR gene itself or in other genes like RBP4 [5] have been discussed in the literature (for review see [6]). It further seems to play a role whether a patient or the affected antecedent is male or female, suggesting that X-linked genes like AR affect TTR amyloidogenesis as well [7]. Furthermore, borderline CAG repeat expansions in the ATXN2 gene are associated with an earlier age of onset [8].
The most prominent physiological function of TTR is transporting thyroxin and retinol binding protein in serum and cerebrospinal fluid (CSF) [9,10,11]. Whereas this function can sufficiently be compensated by other proteins such as thyroglobulin or albumin in serum, TTR is the main thyroxin transporter in the CNS [12]. Due to its proteolytic function and interaction with Aß proteins, a protective role in Alzheimer’s disease has been previously discussed [13] (for review see [14]).
Considering that roughly 90% of TTR is hepatically produced (Fig. 1), liver transplants function similarly to gene therapy. With great success prolonging both span and quality of life, the first liver transplantations were performed in the early 1990s [15,16,17]. Worldwide, 2,181 patients have been registered so far as organ recipients due to ATTRv amyloidosis (http://www.fapwtr.org/ram_fap.htm). Other than surgically, hepatic TTR production can now be targeted by two approved mRNA silencing drugs: patisiran is a small interfering RNA molecule [18] and inotersen an antisense oligonucleotide [19] that both lower serum TTR levels up to 20% of normal, resulting in significant improvement of neuropathy and cardiomyopathy compared to placebo-treated controls. Other than reducing TTR production, the approved substance tafamidis [20, 21], available in daily oral dosages of 20 mg and 61 mg, has been shown to stabilize the TTR tetramer and thereby to decelerate the clinical course including neuropathy and cardiomyopathy, improving overall survival without major side effects.

Physiological TTR production sites. Whereas 90% of TTR is of hepatic origin, other production sites are the choroid plexus and the retina. This becomes of growing importance as these two compartments cannot be sufficiently targeted by currently approved medications. Created with BioRender.com
If treated appropriately, ATTRv amyloidosis might therefore no longer be fatal. This might confront patients and caregivers with new challenges in the future. Out of the currently approved treatments, tafamidis is the only one that crosses the blood–brain barrier, but only minimally [22]. Previous studies have shown that CSF TTR is mainly produced in the choroid plexus [12], and can thereby not be influenced by hepatic TTR mRNA degradation. On the other hand, TTR is least replaceable in the CNS, with little known proteins able to take over its function. This places unique challenges on overall TTR transcript knockdown approaches. Recognizing its neuroprotective role in Alzheimer’s disease [23], it seems paradox that TTR can both prevent and cause dementia.
In this review, we will critically discuss the role of TTR amyloidogenesis in the CNS as well as the subsequent therapeutic challenges. We will summarize the current literature, point towards open questions, and implement an ethical discussion of potential therapeutic long-term effects.
Review
Non-liver-derived transthyretin and associated disease manifestations
To an extent of about 10% [12], TTR is not produced by the liver, but by the choroid plexus and the retinal epithelium (Fig. 1). Protected by the blood–brain barrier (Fig. 2), these central compartments of TTR production are not accessible by large molecules or antisense drugs, neither are they influenced by the effects of liver transplant.

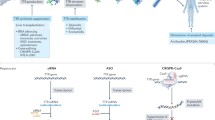
Blood–brain barrier. Schematic depiction of the anatomy and function of the blood–brain barrier. ASO, antisence oligonucleotide; JAM, junctional adhesion molecule; RNAi, ribonucleotide acid interference drug. Created with BioRender.com
Clinically, ocular ATTR deposits cause vitreous or lens opacities, chronic open-angle or neovascular glaucoma, keratoconjunctivits sicca, abnormalities of conjunctival or retinal blood vessels, or optic neuropathy [24, 25]. The clinical spectrum of CNS manifestations is broad, comprising radiculopathy, subarachnoid and intraparenchymal haemorrhage, stroke-like episodes that might as well be explained by seizures, and periods of decreased consciousness [3, 26,27,28].
In the natural course of ATTRv amyloidosis, several variants, including the most frequent, “Portuguese”, mutation p.Val50Met, have CNS or ocular manifestations in their phenotypic spectrum [24]. A predominant eye involvement has been described for the variants p.Arg54Gly, p.Lys55Thr, p.Trp61Leu, p.Tyr89His, and p.Gly83Arg. Leading CNS manifestations have been associated with the variants p.Leu33Pro, p.Asp38Gly, p.Ala45Thr, p.Val50Cys, p.Tyr69Pro, p.Tyr89His, and p.Tyr134Cys. It is interesting to observe that these variants are not the same. It is not understood, however, why one variant increases the liability to one specific, but not to other organ manifestations.
Following liver transplant, Ando et al. described ongoing or new vitreous and leptomeningeal disease manifestations [25]. These were not attributed to wild-type, but to variant TTR, which means that in contrast to heart and other peripheral tissues, these deposits were not formed around previously seeded mutant-derived amyloid, but that there was still new disease activity even after liver transplantation.
Serum TTR is in fact relatively under-represented in the vitreous bodies and CSF, but it is mainly produced by the retina and choroid plexus. In accordance with these observations, patients with iatrogenic ATTRv amyloidosis, meaning recipients of domino-transplanted, fully functioning organs carrying an amyloidogenic TTR mutation, have so far not been observed to develop symptoms other than neuropathy and cardiomyopathy [29,30,31,32], which both result from the presence of unstable TTR in the peripheral circulation. Accordingly, ATTRwt amyloidosis has so far not been described in association with CNS and eye manifestations in the literature.
All three drugs so far approved for the treatment of ATTR amyloidosis (Table 1) have a very limited (tafamidis) or no (patisiran, inotersen) capacity to cross the blood–brain barrier [22]. What we have learned from liver transplanted patients might therefore be applicable in the context of long-term treatment with any of these drugs. Even though CNS and eye manifestations are not the leading phenotype for the majority of TTR variants, longer lifespans enable a different phenotypic pattern under treatment.
The blood–brain barrier and its therapeutic implications
The blood–brain barrier (Fig. 2) consists of non-fenestrated endothelial cells that are closely connected by tight junctions and surrounded by astrocytes and pericytes. Several factors limit the permeability of a systematically delivered therapeutic into the CNS, but none more restrictively than size (less than 4 nm) and charge (preferably uncharged) (reviewed further in [33]). Following these two rules, most therapeutic molecules, including RNA/DNA-based approaches, are both too big and too polar to cross.
The evolution of transient genetic medicine, like RNA interference (RNAi) and antisense oligonucleotides (ASOs) requires either a carrier delivery system or modified RNA or DNA backbone chemistry to increase circulation and half-life [34, 35]. Unmodified RNA or DNA sequences used in RNAi or ASOs tend to quickly exit circulation through renal filtration and limit their intracellular survival against endo- and exo-nucleases [34, 36]. Bearing this in mind, it is patently clear why the most efficacious molecular adaptations made to these strategies are counterproductive for their systemic and targeted distribution. For this reason, the most obvious solution is direct administration into the CNS via intrathecal delivery, a method adapted for the treatment of aggressively progressive disorders like spinal muscular atrophy (SMA), amyotrophic lateral sclerosis (ALS), and Huntington’s Disease [37]. Currently, nusinersen is the only intrathecally delivered ASO approved by the FDA and EMA [38, 39], however, several others are in trial for the previously mentioned disorders.
In ATTRv amyloidosis, the small molecule tafamidis is the only approved therapeutic that can cross the blood–brain barrier, however, no more than 1.5% of the plasma circulating drug actually reach the cerebrospinal fluid [22]. Like tafamidis, the catechol-O-methyltransferase (COMT) inhibitor tolcapone, approved for the treatment of Parkinson’s disease, has a TTR stabilizing effect [40] with a greater capacity to cross the blood–brain barrier [41, 42]. Relevant limitations of this drug are, however, the short half-life and the potential side effects including liver failure.
Using Watson–Crick base pairing, antisense oligonucleotide and RNA-based strategies have historically elicited knockdown mechanisms as plausible treatment approaches, especially for dominantly inherited disorders. This is the case for both patisiran, a double stranded RNA-interference (RNAi) therapeutic, and inotersen, a single-stranded antisense oligonucleotide [43, 44]. While the mechanism of action differs, both aim to indiscriminately reduce wildtype and mutant TTR RNA transcripts by targeting a 3’-UTR sequence in the pre-mRNA, ultimately reducing aggregate formation.
Patisiran uses the well-established siRNA strategy of binding to a target RNA transcript and recruiting the RNA-induced silencing complex (RISC) to cleave the transcript, exposing it to exonucleases, subsequent degradation and eventual clearance, but allowing the siRNA to continue binding to other target transcripts. Packaged into a lipid nanoparticle delivery system targeted to the liver, it has proven efficacious in knocking down TTR transcripts in the most relevant organ for TTR production and remaining active for weeks with mitigated secondary and no documented off-target effects. However, it fails to address the ongoing need for targeting production of TTR in the choroid plexus, where continued transcription of amyloidogenic mutant alleles progressively endanger susceptible tissues, including CNS and eyes. Direct and titrated administration of patisiran into the CNS has yet to be published, potentially due to the perceived negligible advantages to repeated intrathecal delivery to tissue not primarily responsible for the majority of TTR production. Underlining this logic is the expressed improvement of patients in regards to their neuropathy and cardiomyopathy [18, 45, 46], both not requiring the drug to cross the blood–brain barrier. However, an intrathecal route of administration is currently under investigation for siRNA therapeutics targeting neurodegenerative diseases like Alzheimer’s or Huntington’s disease. For these indications, phase 1 clinical trials are expected to start recruiting soon (information provided by Alnylam, manufacturer of patisiran).
Inotersen uses a short 20 bp chemically modified DNA sequence to bind a complimentary section of the TTR pre-mRNA 3′-UTR, leading to the recruitment of RNAse H1, a nuclear ribonuclease recognizing DNA-RNA duplexes, and cleavage of the target transcript. Similarly to patisiran, cleavage and clearance of the target transcript permit the ASO to effectively reduce TTR translation. The ASO half-life and resistance to intracellular degradation is owed to its charged backbone and 2’-O-methoxyethyl substitutions in the ribose sugar as well as the phosphorothioate substitutions to the oxygen in the phosphate group. These modifications confer both stability and affinity to a target transcript but impede its penetrance to the CNS [47]. As mentioned above, nusinersen, another ASO drug, has already been approved for intrathecal treatment of spinal muscular atrophy [38, 39]. The mechanism used here is not RNA degradation, but expression regulation through splice modifications. However, the fact that another drug of the ASO family has already been shown to be safely administered to the CSF gives hope for future developments in other indications.
TTR knockdown in CNS: potential limitations related to TTR-function
If the route of administration was no longer a limitation for CNS TTR knockdown, would it then be its irreplaceable function?
In the CSF, TTR represents 20% of total proteins, with levels ranging between 1.5 and 2.5 mg/dL [48]. Besides the choroid plexus, neurons themselves might contribute to the central TTR production, which has been discussed as a response to neuronal stress. The amount of this effect has not been quantified to date. Several animal studies suggested a neuroprotective role of TTR in the context of peripheral nerve injury [49] or cerebral ischemia [50]. TTR stability seemed to be a positive predictor for favorable outcome in a large screening study on strokes in young adults [51]. In several studies on Alzheimer’s disease (AD), CSF TTR levels were shown to be reduced [52, 53], however, this does not seem to be specific for AD [54] nor consistently reproduced by other studies [55]. How exactly TTR interacts with Aβ and attenuates its toxicity, is not fully understood to date (for review see [14]). The proteolytic cleavage of Aβ fibrils by TTR [56] is one of the hypothesized mechanisms. As shown in cell culture, the uptake of Aβ protein correlated with TTR stability [44]. The overexpression of TTR improved cognition and reduced the neuropathological disease load in mice [57], whereas TTR knock-out led to an accelerated disease course [57]. Beyond AD disease models, TTR knockdown seemed to result in reduced memory function in otherwise healthy mice [58]. Other discussed neuroprotective roles of TTR comprise neuro-regeneration and neuropeptide homeostasis [9].
While some of these TTR functions are closely related to its role in transporting retinol binding protein [58], others might require a direct interaction between TTR and other proteins. These considerations might become important in a (iatrogenic) loss-of-function situation. While the former could be partially replaced by vitamin A substitution, which is already an established requirement under antisense drug medication [18, 19], the latter needs to be further investigated in order to fully assess and address potential risks.
Potential answers/strategies
In summary, we encounter two major challenges when trying to treat CNS ATTRv amyloidosis:
-
1.
How to target the CNS with the lowest possible toxicity and application burden for the patient?
-
2.
How to selectively reduce the expression of the mutant, but not of the essential wildtype allele?
Therefore, the next intuitive frontier of genetic medicine is both optimizing tissue specific distribution and molecular precision.
Increasing tissue specificity is an important step to reduce overall toxicity. For the indication ATTRv amyloidosis, several translation modifying drugs (vutrisiran, eplontersen) are currently in phase 3 clinical trials that due to a modification with N-acetylgalactosamine (GalNAc) have an enhanced delivery to the liver [59, 60]. Unfortunately, there is no such selective, receptor mediated uptake system for the CNS that could be exploited to date.
Targeted delivery of intrathecally administered therapeutics has just recently begun to play a role in ATTRv amyloidosis: Alnylam Pharmaceuticals has conducted pre-clinical studies, using murine models for targeted ocular TTR transcript knockdown (Nair_OTS2018 (alnylam.com)). Achieving sustained circulation through CSF and increased tissue specific penetrance overcomes the most challenging barriers in CNS specific delivery.
Targeting allele-specific RNA transcripts comes with a unique sequence and gene dependent challenge as distinguishing between the two transcripts depends on how specifically the complimentary therapeutic can bind to one, but not the other allele. For the purpose of CNS-delivered genetic therapeutics, an allele- specific approach would permit for wt TTR to remain the primary thyroxin and retinol binding protein transporter, while significantly diminishing the mutant transcripts. Molecular precision can be thought as an interplay between sequence specificity for optimal siRNA or ASO binding and increased therapeutic half-life for continued transcript clearance with mitigated intracellular toxicity. Calibrating appropriate therapeutic dosing is further complicated by the variability in DNA/RNA backbone modifications made to the therapeutic itself. Most FDA-approved DNA/RNA therapies have a handful of tried-and-true substitutions conferring stability and increased specificity, but the combination in which they are used (i.e. chimeric, gapmer, and stereopure) are a niche titrating step as important to molecular precision as to toxicity.
Currently, one limitation to sequence-specific therapeutics might be that clinical trial pipelines eventually aiming at drug approval require every modification of a certain drug to undergo the full process of clinical testing, including phase 2 and 3 trials. Considering that about 130 amyloidogenic variants are known todate, it seems impossible (not only financially, but also in respect of low case numbers) to overcome these regulatory limitations for every single TTR mutation. Other than targeting the respective variant itself, however, allele-specific knock-down could also mean to target frequent polymorphisms located in cis with the mutated allele. This would require a more detailed genetic testing, including zygosity studies, but enable to optimize the development pipeline focusing on just a few frequent variations.
The CRISPR/Cas9 system, the subject of the 2020 Nobel Prize in chemistry, has garnered fame and support for its potential in permanently editing genomic DNA. This technique has the advantages 1) to reduce the application burden by permanent editing and 2) to specifically rewrite the causative DNA mutations. Clinical trials for Sickle cell disease and ß-Thalassemia, whose causal mutations are found in the ß-hemoglobin subunit gene HBB, have recently shown CRISPR/Cas9 to reduce production of a γ-globin repressor [61, 62]. For ATTRv amyloidosis, a Phase I open-label, multi-center study is currently underway in the United Kingdom to evaluate the safety, tolerability, and efficacy of NTLA-2001 sponsored by Intellia Therapeutics and Regeneron Pharmaceuticals [63]. Administered intravenously, with no ability to cross the blood–brain barrier, the CRISPR/Cas9 system travels predominantly to the liver in lipid nanoparticles. Using a single guide RNA, the system is designed to edit the TTR gene and completely disrupt its transcription by essentially creating a second knockout mutation. In the six eligible participants, none showed severe adverse events and all had a drastic decrease in serum TTR concentration, with those receiving the highest dose (0.3 mg/kg) experiencing an over 80% reduction 28 days after infusion [63]. Participants will be continually monitored and followed for 24 months after the initial infusion to determine sustained knockdown. While this relieves the primary burden of systemic disease, patients may continue to present with CNS and ocular symptoms as it will not affect TTR transcription other than hepatic.
Considering the amount of promising future treatment options for TTR, combination therapy may be the best tailored option using TTR stabilizers with the potential to cross the blood–brain barrier, intrathecally administered RNAi and ASOs, and genome editing strategies in a precision medicinal approach to eliminate the root cause.
Are we creating a new phenotype? Ethical aspects
The availability of the aforementioned treatments for ATTRv amyloidosis is an undoubtedly positive development in terms of the possibility of increased lifespan and reduction of symptoms. Nevertheless, these treatments are not a cure for ATTRv amyloidosis, and raise myriad questions: What will happen when lifespans increase, but central amyloid production continues? Will we be able to mitigate neuropathy and cardiomyopathy, while treated patients have to face new issues including seizures, dementia, and blindness? What long-term impact do these treatments have on patients’ and their caregivers’ well-being and quality of life? Is this impact potentially greater than that of the initial phenotype? While the answers to some of these questions rely solely on clinical data that is not yet available, some raise specific ethical issues that we aim to illustrate in the following paragraphs.
-
Benefits vs. harms
From an ethical standpoint, it is important to note that the risk of generating a new phenotype of a disease through treatment is not new, and has already been discussed in fields such as HIV, transplants [64], and, specifically in neurology, deep brain stimulation (DBS) for Parkinson’s Disease (PD) [65], and new treatments for amyotrophic lateral sclerosis (ALS) [66]. In all of these cases, the benefits of the treatment are evident and have been amply documented, as are the risks that come with an increased lifespan. Indeed, this discussion is likely to take place in an increasing number of clinical scenarios, as new treatment options increase patients’ lifespans, enabling known diseases to reach advanced stages of unknown severity [67]. In some cases, such as the one at hand, it is not only the severity of known symptoms, but the possibility of the emergence of new symptoms that is worrisome. From liver-transplanted ATTRv amyloidosis patients, we have learned that CNS and eye symptoms cannot be stopped if the choroid plexus and retina continue to produce mutant TTR. In the long-term, carriers of TTR mutations that are not specifically associated with predominant central involvement have survived long enough to eventually develop cerebral amyloid angiopathy or other CNS and eye manifestations. However, these risks and concerns must be weighed against the reality of ATTRv amyloidosis without treatment, and the evidence for currently available treatments. Thus, the main ethical questions we must ask are: Do the benefits of treatment outweigh the harms? Can these negative aspects of the new phenotype be treated with currently available agents, or is it foreseeable that they will be in the future? The currently approved medications cannot sufficiently overcome this problem. Should research focus shift to drugs that enforce TTR knockdown in the CNS and vitreous bodies as well? Or would that deprive these compartments from the protective roles of the TTR protein that might not play a relevant role in serum? Certain forms of treatment for ATTRv amyloidosis may come with a degree of iatrogenic harm, not only due to the emergence of a new phenotype, but also due to the currently limited perspective for successful treatment options that do not result in further harms resulting from the impairment of TTR functions. Our main focus now should be on the development of agents that can help prevent or treat the foreseeable symptoms of a possible new phenotype.
-
Informed consent and treatment plan
In order to ethically implement treatment options that may give rise to a new phenotype of a disease, we must include all relevant information in the informed consent process, enabling an autonomous decision. An optimal clinical scenario would incorporate a written consent form that encompasses all relevant information, including: (a) a detailed description of the treatment and its many significant potential benefits, (b) the possibility of specific new symptoms, and c) a drafted future treatment plan. The aim of incorporating these aspects into informed consent discussions is not only to allow for careful consideration of both the potential benefits and consequences of treatment in order to decide whether to undergo the treatment at all (is the risk of blindness, dementia or epilepsy, among others, one that the patient is unwilling to accept when considering the benefit of the treatment?), but also to discuss what impact these risks would have on the patient’s perceived quality of life. Increasing patients’ lifespan while increasing their chances of blindness or dementia may mean that they will need care and may not be able to fulfill their expectations and values of a meaningful life or acceptable levels of well-being. It is also important to consider the risk of dementia and its effect on the capacity to consent to future treatments that may have positive effects on ATTRv amyloidosis. In order to safeguard patient autonomy, the discussion on whether to undergo treatment ought not be separated from the discussion of the patient’s wishes and values regarding their future treatment. Because of this, it is advisable to explicitly discuss drafting a future treatment plan, ideally in the form of advance directives, during the informed consent process.
Nevertheless, it is important that we underscore the proven benefits of the mentioned treatments during the process of informed consent: it is arguably as much a disservice to patient autonomy to insufficiently address risks as it is to overstate them or insufficiently address benefits. These scenarios could result, at worst, in a refusal of treatment due to a lack of hope in treatment, or, at best, to poor compliance.
-
Evidence-based decisions without evidence?
Ethical clinical practice ideally ought to be based on data from well-designed randomized controlled clinical trials. This poses a challenge in rare diseases such as ATTRv amyloidosis, or treatment options that are considered “last resorts”, where patient numbers are lower and outcomes are less likely to be standardized. In addition, when effective treatment options are available, the ethical justification for placebo controlled trials where conditions for equipoise are not met becomes challenging at best. Due to the limited availability of data from clinical trials, authors have suggested the systematic collection of patient data [66] into rare disease registries with standardized treatment and outcome measures. The need for and potential benefits of these registries is highlighted in phenotype-modifying cases such as the one at hand, where a degree of uncertainty in certain aspects of treatment outcome is currently inevitable. As an example, the Transthyretin Amyloidosis Outome Survey (THAOS) has been systematically collecting demographic, clinical, and genetic data in more than 1400 ATTRv amyloidosis cases [68]. It does, however, not systematically assess the phenotypic changes under all available treatment options. This is a major limitation for the current question.
-
Stakeholder perspectives
As mentioned above, liver-transplanted ATTRv amyloidosis patients present continued progression of CNS and eye symptoms if the choroid plexus and retina continue to produce mutant TTR. To the best of our knowledge, there are no published works focusing on the perspectives of patients and their caregivers on the development of new symptoms and their consequences for their well-being and quality of life. The same can be said on works exploring healthcare professionals’ experiences in this context and their attitudes towards treatment. Future practice and regulation in this area would greatly benefit from such input, and most importantly, could be extremely helpful in patients’ decision-making process.
Conclusions
With the liver in the center of pathophysiology, ATTR amyloidosis has become a model disease for mechanism-based treatment approaches. Compared to other systemic or non-systemic diseases affecting the nervous system, the possibility to reduce the hepatic TTR production and thereby stop disease progression in almost all the other organs and tissues is a great advantage, given that various routes of administration are effective to target the liver. For the about 10% of extra-hepatic TTR production, there is, however, no good treatment strategy available to stop progression. It is conceivable that with longer survival, even TTR variants that have so far not primarily been associated with CNS and ocular disease manifestations might lead to clinically relevant amyloid angiopathy and vitreous opacities. That means that cerebral hemorrhage, dementia, and blindness might be the new phenotype of ATTRv amyloidosis in the long-term.
In the near future, it might be realistic to expect patients will receive RNAi combination therapies, one targeting primary TTR systemic production and the other targeting CNS specific TTR. Alternatively, physicians might be able to tailor either treatment strategy dependent on disease course and presenting symptoms.
To overcome the expected side effects of unspecific TTR knockdown in the CNS, an allele specific approach would be promising to selectively reduce the expression of the mutant, but not of the wildtype. Using the CRISPR gene editing technique, first steps are being made in that direction for ATTRv amyloidosis now, however, they have been, again, limited to hepatic TTR production so far.
In this review, we addressed and discussed two important questions that are both related to TTR function and dysfunction in the nervous system. First, we pointed out that the currently available treatment approaches do not target ATTR amyloid depositing in the eyes or CNS, which will eventually increase the impact of long-term “natural course” complications such as cerebral hemorrhage, dementia, and blindness with prolonged life-expectancies. Secondly, we emphasized that TTR might be of greater functional relevance in CSF than in serum, and that simple vitamin A substitution might not be sufficient to fully replace its neuroprotective effects. Taken together, central TTR knockdown might be helpful or even needed to stop CNS ATTRv amyloidosis, which is, without doubt, a dreadful and life-limiting condition. This can, on the other hand, potentially favor the development of dementia as well. TTR stabilization could be a compromise – at least in the function of bridging until disease progression eventually requires protein knockdown. This requires, however, a higher percentage of CSF availability.
Lastly, we considered the main ethical concerns raised:
-
The generation of a new phenotype of ATTRv amyloidosis as a potential iatrogenic harm that must be weighed against the undoubted benefits of treatments
-
The crucial importance of good practices in informed consent with detailed descriptions of both potential benefits and harms and the incorporation of a) a written informed consent form, and b) the discussion of advance directives into the process of informed consent
-
The challenges of a paucity of data from randomized controlled trials and the importance of rare disease registries
-
The need for stakeholder perspectives to inform future clinical practice and guidelines.
Great success has been made in the past decade turning ATTRv amyloidosis into a treatable disease and offering new perspectives to patients and families. To further improve these perspectives and to avoid new dreadful disease manifestations, many open questions will still have to be addressed in the future.
Availability of data and materials
Not applicable.
Abbreviations
- ALS:
-
Amyotrophic lateral sclerosis
- ASO:
-
Antisense oligonucleotide
- ATTRv amyloidosis:
-
Hereditary transthyretin amyloidosis
- ASGPR:
-
Asialoglycoprotein receptor
- CNS:
-
Central nervous system
- COMT:
-
Catechol-O-methyltransferase
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- CSF:
-
Cerebrospinal fluid
- DBS:
-
Deep brain stimulation
- DNA:
-
Desoxy-ribonucleic acid
- EMA:
-
European Medicines Agency
- FDA:
-
Food and Drug Administration
- GalNac:
-
N-Acetylgalactosamine
- JAM:
-
Junctional adhesion molecule
- PD:
-
Parkinson’s disease
- RISC:
-
RNA-induced silencing complex
- RNA:
-
Ribonucleic acid
- siRNA:
-
Small interfering RNA
- SMA:
-
Spinal muscular atrophy
- THAOS:
-
Transthyretin Amyloidosis Outome Survey
- TTR:
-
Transthyretin
- UTR:
-
Untranslated region
References
Planté-Bordeneuve, V., & Said, G. (2011). Familial amyloid polyneuropathy. Lancet Neurology, 10(12), 1086–1097.
Westermark, P., et al. (1990). Fibril in senile systemic amyloidosis is derived from normal transthyretin. Proceedings of the National Academy of Sciences of the United States of America, 87(7), 2843–2845.
Maia, L., et al. (2015). CNS involvement in V30M transthyretin amyloidosis: Clinical, neuropathological and biochemical findings. Journal of Neurology, Neurosurgery and Psychiatry, 86(2), 159.
Beirao, J. M., et al. (2015). Ophthalmological manifestations in hereditary transthyretin (ATTR V30M) carriers: A review of 513 cases. Amyloid, 22(2), 117–122.
Soares, M. L., et al. (2005). Susceptibility and modifier genes in Portuguese transthyretin V30M amyloid polyneuropathy: Complexity in a single-gene disease. Human Molecular Genetics, 14(4), 543–553.
Dohrn, M. F., et al. (2021). Targeting transthyretin-Mechanism-based treatment approaches and future perspectives in hereditary amyloidosis. Journal of Neurochemistry, 156(6), 802–818.
Santos, D., et al. (2016). Variants in RBP4 and AR genes modulate age at onset in familial amyloid polyneuropathy (FAP ATTRV30M). European Journal of Human Genetics, 24(5), 756–760.
Santos, D., et al. (2019). Large normal alleles of ATXN2 decrease age at onset in transthyretin familial amyloid polyneuropathy Val30Met patients. Annals of Neurology, 85(2), 251.
Liz, M. A., et al. (2010). Aboard transthyretin: From transport to cleavage. IUBMB Life, 62(6), 429–435.
Woeber, K. A., & Ingbar, S. H. (1968). The contribution of thyroxine-binding prealbumin to the binding of thyroxine in human serum, as assessed by immunoadsorption. The Journal of Clinical Investigation, 47(7), 1710–1721.
Smith, F. R., Raz, A., & Goodman, D. S. (1970). Radioimmunoassay of human plasma retinol-binding protein. The Journal of Clinical Investigation, 49(9), 1754–1761.
Herbert, J., et al. (1986). Transthyretin: A choroid plexus-specific transport protein in human brain. The 1986 S. Weir Mitchell award. Neurology, 36(7), 900–911.
Li, X., et al. (2011). Neuronal production of transthyretin in human and murine Alzheimer’s disease: Is it protective? Journal of Neuroscience, 31(35), 12483.
Li, X., & Buxbaum, J. N. (2011). Transthyretin and the brain re-visited: Is neuronal synthesis of transthyretin protective in Alzheimer’s disease? Molecular Neurodegeneration, 6, 79.
Holmgren, G., et al. (1991). Biochemical effect of liver transplantation in two Swedish patients with familial amyloidotic polyneuropathy (FAP-met30). Clinical Genetics, 40(3), 242–246.
Holmgren, G., et al. (1993). Clinical improvement and amyloid regression after liver transplantation in hereditary transthyretin amyloidosis. Lancet, 341(8853), 1113–1116.
Ericzon, B.-G., et al. (2015). Liver transplantation for hereditary transthyretin amyloidosis: After 20 years still the best therapeutic alternative? Transplantation, 99(9), 1847–1854.
Adams, D., et al. (2018). Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. The New England Journal of Medicine, 379(1), 11–21.
Benson, M. D., et al. (2018). Inotersen treatment for patients with hereditary transthyretin amyloidosis. New England Journal of Medicine, 379(1), 22–31.
Coelho, T., et al. (2012). Tafamidis for transthyretin familial amyloid polyneuropathy: A randomized, controlled trial. Neurology, 79(8), 785–792.
Maurer, M. S., et al. (2018). Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. New England Journal of Medicine, 379(11), 1007–1016.
Monteiro, C., et al. (2018). Cerebrospinal fluid and vitreous body exposure to orally administered tafamidis in hereditary ATTRV30M (p. TTRV50M) amyloidosis patients. Amyloid, 25(2), 120–128.
Gião, T., et al. (2020). Undiscovered roles for transthyretin: from a transporter protein to a new therapeutic target for Alzheimer’s disease. International Journal of Molecular Sciences, 21(6), 2075.
Dammacco, R., et al. (2020). Amyloidosis and ocular involvement: An overview. In Seminars in ophthalmology.
Ando, E., et al. (1997). Ocular manifestations of familial amyloidotic polyneuropathy type I: Long term follow up. British Journal of Ophthalmology, 81(4), 295–298.
Ushiyama, M., Ikeda, S., & Yanagisawa, N. (1991). Transthyretin-type cerebral amyloid angiopathy in type I familial amyloid polyneuropathy. Acta Neuropathologica, 81(5), 524.
Herrick, M., et al. (1996). Massive leptomeningeal amyloidosis associated with a Val30Met transthyretin gene. Neurology, 47(4), 988.
Sakashita, N., et al. (2001). Familial amyloidotic polyneuropathy (ATTR Val30Met) with widespread cerebral amyloid angiopathy and lethal cerebral hemorrhage. Pathology International, 51(6), 476.
Vollmar, J., et al. (2018). Progression of transthyretin (TTR) amyloidosis in donors and recipients after domino liver transplantation—A prospective single-center cohort study. Transplant International, 31(11), 1207–1215.
Lladó, L., et al. (2010). Risk of transmission of systemic transthyretin amyloidosis after domino liver transplantation. Liver Transplantation, 16(12), 1386.
Adams, D., et al. (2011). Symptomatic and proven de novo amyloid polyneuropathy in familial amyloid polyneuropathy domino liver recipients. Amyloid, 18(sup1), 174–177.
Stangou, A. J., Heaton, N. D., & Hawkins, P. N. (2005). Transmission of systemic transthyretin amyloidosis by means of domino liver transplantation. New England Journal of Medicine, 352(22), 2356.
Daneman, R., & Prat, A. (2015). The blood–brain barrier. Cold Spring Harbor Perspectives in Biology, 7(1), a020412.
Scoles, D. R., Minikel, E. V., & Pulst, S. M. (2019). Antisense oligonucleotides: A primer. Neurology Genetics, 5(2), 323.
Kanasty, R., et al. (2013). Delivery materials for siRNA therapeutics. Nature Materials, 12(11), 967–977.
Whitehead, K. A., Langer, R., & Anderson, D. G. (2009). Knocking down barriers: Advances in siRNA delivery. Nature Reviews. Drug Discovery, 8(2), 129–138.
Scoles, D. R., & Pulst, S. M. (2018). Oligonucleotide therapeutics in neurodegenerative diseases. RNA Biology, 15(6), 707.
Finkel, R. S., et al. (2017). Nusinersen versus sham control in infantile-onset spinal muscular atrophy. New England Journal of Medicine, 377, 1723–1732.
Mercuri, E., et al. (2018). Nusinersen versus sham control in later-onset spinal muscular atrophy. New England Journal of Medicine, 378(7), 625–635.
Sant’Anna, R., et al. (2016). Repositioning tolcapone as a potent inhibitor of transthyretin amyloidogenesis and associated cellular toxicity. Nature Communications, 7(1), 1–13.
Russ, H., et al. (1999). Detection of tolcapone in the cerebrospinal fluid of parkinsonian subjects. Naunyn-Schmiedeberg’s Archives of Pharmacology, 360(6), 719–720.
Pinheiro, F., et al. (2020). Tolcapone, a potent aggregation inhibitor for the treatment of familial leptomeningeal amyloidosis. The FEBS Journal, 288, 310–324.
Kristen, A., et al. (2019). Patisiran, an RNAi therapeutic for the treatment of hereditary transthyretin-mediated amyloidosis. Neurodegenerative Disease Management, 9(1), 5.
Mathew, V., & Wang, A. K. (2019). Inotersen: New promise for the treatment of hereditary transthyretin amyloidosis. Drug Design, Development and Therapy, 13, 1515.
González-Duarte, A., et al. (2020). Analysis of autonomic outcomes in APOLLO, a phase III trial of the RNAi therapeutic patisiran in patients with hereditary transthyretin-mediated amyloidosis. Journal of Neurology, 267(3), 703–712.
Solomon, S. D., et al. (2019). Effects of patisiran, an RNA interference therapeutic, on cardiac parameters in patients with hereditary transthyretin-mediated amyloidosis: Analysis of the APOLLO study. Circulation, 139(4), 431–443.
Bennett, C. F., et al. (2017). Pharmacology of antisense drugs. Annual Review of Pharmacology and Toxicology, 57, 81–105.
Weisner, B., & Roethig, H.-J. (1983). The concentration of prealbumin in cerebrospinal fluid (CSF), indicator of CSF circulation disorders. European Neurology, 22(2), 96–105.
Fleming, C. E., Saraiva, M. J., & Sousa, M. M. (2007). Transthyretin enhances nerve regeneration. Journal of Neurochemistry, 103(2), 831–839.
Santos, S. D., et al. (2010). CSF transthyretin neuroprotection in a mouse model of brain ischemia. Journal of Neurochemistry, 115(6), 1434–1444.
Gao, C., et al. (2011). Serum prealbumin (transthyretin) predict good outcome in young patients with cerebral infarction. Clinical and Experimental Medicine, 11(1), 49–54.
Riisøen, H. (1988). Reduced prealbumin (transthyretin) in CSF of severely demented patients with Alzheimer’s disease. Acta Neurologica Scandinavica, 78(6), 455.
Serot, J., et al. (1997). Cerebrospinal fluid transthyretin: Aging and late onset Alzheimer’s disease. Journal of Neurology, Neurosurgery and Psychiatry, 63(4), 506.
Ranganathan, S., et al. (2005). Proteomic profiling of cerebrospinal fluid identifies biomarkers for amyotrophic lateral sclerosis. Journal of Neurochemistry, 95(5), 1461.
Schultz, K., et al. (2010). Transthyretin as a potential CSF biomarker for Alzheimer’s disease and dementia with Lewy bodies: Effects of treatment with cholinesterase inhibitors. European Journal of Neurology, 17(3), 456.
Silva, C., et al. (2017). Transthyretin neuroprotection in Alzheimer’s disease is dependent on proteolysis. Neurobiology of Aging, 59, 10.
Buxbaum, J. N., et al. (2008). Transthyretin protects Alzheimer’s mice from the behavioral and biochemical effects of Aβ toxicity. Proceedings of the National Academy of Sciences of the United States of America, 105(7), 2681–2686.
Brouillette, J., & Quirion, R. (2008). Transthyretin: A key gene involved in the maintenance of memory capacities during aging. Neurobiology of Aging, 29(11), 1721–1732.
Habtemariam, B. A., et al. Single dose pharmacokinetics and pharmacodynamics of transthyretin targeting GalNAc-siRNA conjugate, vutrisiran, in healthy subjects. Clinical Pharmacology & Therapeutics.
Khella, S., et al. (2020). Rationale and design of NEURO-TTRansform, a phase 3 study to evaluate the efficacy and safety of AKCEA-TTR-LRx (ION-682884) in Patients with hereditary transthyretin-mediated amyloid polyneuropathy (2240). AAN Enterprises.
Esrick, E. B., et al., Post-transcriptional genetic silencing of BCL11A to treat sickle cell disease. New England Journal of Medicine.
Frangoul, H., et al. (2021). CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. New England Journal of Medicine, 384(3), 252–260.
Gillmore, J. D., et al. (2021). CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. New England Journal of Medicine, 385(6), 493–502.
Chapman, J. R. (2011). The consequences of successful transplantation. Lancet, 378(9800), 1357–1359.
Gilbert, F., & Lancelot, M. Incoming ethical issues for deep brain stimulation: when long-term treatment leads to a'new form of the disease'. Journal of Medical Ethics, p. medethics-2019-106052.
Schorling, D. C., Pechmann, A., & Kirschner, J. (2020). Advances in treatment of spinal muscular atrophy-new phenotypes, new challenges, new implications for care. Journal of Neuromuscular Disease, 7(1), 1.
Weaver, F. M., et al. (2017). Survival in patients with Parkinson’s disease after deep brain stimulation or medical management. Movement Disorders, 32(12), 1756–1763.
Coelho, T., et al. (2013). THAOS-The transthyretin amyloidosis outcomes survey: Initial report on clinical manifestations in patients with hereditary and wild-type transthyretin amyloidosis. Current Medical Research and Opinion, 29(1), 63–76.
Acknowledgements
We thank Teresa Coelho and Stephan Züchner for inspiring mentoring.
Funding
This work has been supported by the Interdisciplinary Center of Clinical Research Aachen (IZKF TN1-9, 532009). MFD is currently receiving a scholarship by the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG) (Grant No. DO 2386/1-1).
Author information
Authors and Affiliations
Contributions
MFD and EH conceptualized the manuscript. MFD, JM, and KROD drafted the manuscript. All authors critically reviewed and edited the manuscript. MFD and JM contributed equally. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
MFD received financial reimbursement for consulting and advisory board activities and travel support to attend scientific meetings by Akcea Therapeuticals Inc., Alnylam Pharmaceuticals Inc., and Pfizer Pharmaceuticals. MFD further received research funding by Pfizer Pharmaceuticals (ASPIRE 2018). JM has no conflicts. KROD has received speaker fees from Eisai GmBH. EH received financial reimbursement for consulting and advisory board activities and/or travel support by Akcea, Alnylam, and Pfizer.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dohrn, M.F., Medina, J., Olaciregui Dague, K.R. et al. Are we creating a new phenotype? Physiological barriers and ethical considerations in the treatment of hereditary transthyretin-amyloidosis. Neurol. Res. Pract. 3, 57 (2021). https://doi.org/10.1186/s42466-021-00155-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42466-021-00155-8