
Abstract
Background
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a severe hereditary channelopathy characterized by the presence of ventricular arrhythmias triggered by adrenergic stimuli, usually diagnosed in the first two decades of life. Genetic variants in the cardiac ryanodine receptor gene are the most frequently occurring that cause an increase in intracellular calcium concentration and thus induce ventricular arrhythmias due to a delayed after depolarisation-induced triggered activity.
Case presentation
We present the case of a 74-year-old male, a regular athlete with no relevant family history who suffered from sinus dysfunction and frequent premature ventricular complexes with no symptoms. A treadmill test revealed severe polymorphic ventricular arrhythmias which led to the suspicion of CPVT. A genetic study was undertaken, and it identified a rare genetic variant in the RYR2 gene which was possibly associated with its development in heterozygosity: c.14465G > A, p.Arg4822His. While evaluating the co-segregation, we observed that most of his relatives exhibit polymorphic ventricular arrhythmias with exertion without symptoms and carried the same variant.
Conclusions
We described, for the first time, the clinical characteristics and co-segregation of a family diagnosed with CPVT secondary to a little-known genetic variant of the RYR2 gene. It is a variant that, in our case study, suggests an association with a very good prognosis.
Similar content being viewed by others


Background
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a hereditary channelopathy characterized by the presence of polymorphic ventricular arrhythmias and typically bidirectional ventricular tachycardias (VT) triggered by adrenergic stimuli. It usually occurs in young people with a structurally normal heart and an anodyne electrocardiogram (ECG) at rest. It is a disease with a low prevalence but, at the same time, has high mortality [1].
Genetic variants in the cardiac ryanodine receptor gene (RYR2)—which are transmitted with an autosomal dominant inheritance pattern—are the most frequently occurring (approximately 65% of cases) and cause an increase in intracellular calcium concentration. Thus, they induce ventricular arrhythmias due to a delayed after depolarisation-induced triggered activity [1].
Case presentation
We present the case of a family diagnosed with CPVT in which we found a rare genetic variant in the RYR2 gene.
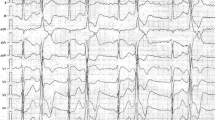
The index case is a 74-year-old male, a regular athlete with no relevant family history. He suffered from sinus bradycardia and frequent premature ventricular complexes (PVCs) with no symptoms. The echocardiography did not reveal any abnormality. A treadmill test was conducted to evaluate the chronotropic response, and PVCs were observed at rest. Initially, they became frequent, with bigeminy and doublets of polymorphic PVCs from stage 1 of the BRUCE protocol, and then episodes of non-sustained and incessant polymorphic VT appeared before completing stage 2 of the Bruce protocol that were temporarily bidirectional (Fig. 1). The study was completed with a coronary angiography and a cardiac MRI in which no significant observations were made.

12-lead electrocardiographic recordings of the index case obtained during the treadmill test in which polymorphic and bidirectional ventricular tachycardias are observed
Treatment with beta-blockers was commenced without observing new episodes of ventricular arrhythmias during admission. However, owing to limitations caused by the significant sinus dysfunction with sinus pauses and episodes of nodal rhythm, and in view of increasing doses, a permanent pacemaker was implanted before the patient’s hospital discharge.
Given the suspicion of a CPVT, a genetic study was undertaken using the Next-Generation Sequencing (NGS) method. The panel included 77 genes, and it identified a likely pathogenic variant in the RYR2 gene which was possibly associated with its development in heterozygosity: c.14465G > A, p.Arg4822His. While evaluating the co-segregation through a targeted genetic test, we observed that, out of the patient’s three children, only one had a negative genetic result and did not exhibit ventricular arrhythmias. The remaining two were positive for the same variant, and they presented episodes of polymorphic ventricular arrhythmias with exertion. As for his three siblings, his two sisters presented a positive genetic result and ventricular arrhythmias with exertion. The other brother could not be studied, but a treadmill test he had previously undertaken for another condition that had revealed polymorphic PVCs and non-sustained VTs. Regarding the rest of the relatives of the index patient, the descendants of the sisters were also studied, as recorded in the genealogical tree (Fig. 2). None of them showed symptoms or presented any echocardiographic abnormalities. Positive cases were treated with nadolol; they remain asymptomatic after more than 3 years of follow-up, with a significant reduction in ventricular arrhythmias in subsequent treadmill tests.

Genealogical tree of the family. The arrow indicates the proband, and the asterisks denote the patients who have been evaluated by our medical team
Discussion
CPVT is usually diagnosed in the first two decades of life, with a higher risk of sudden death the earlier symptoms appear. Late-onset cases, such as in patients aged over 40 years, have been described in the existing literature and have been associated with a better prognosis [2]. This family represents a rare case of diagnosis in adulthood, thereby emphasizing the importance of maintaining a high degree of suspicion for the effective and early diagnosis of this entity. It is also worth focusing on the absence of symptoms in our patients, which is consistent with previous family studies that have observed a high proportion of asymptomatic patients [3]. The reason is unknown and warrants further study to elucidate the genetic modifiers responsible for the variability in phenotypic expression.
Another unique aspect of this case study was that we arrived at this family’s diagnosis as a result of the study of a sinus dysfunction in the index case. RYR2 mutations have been associated with sinus dysfunction and atrial tachyarrhythmias since the ryanodine receptor is also present in the atrial myocardium and the pacemaker cells [4]. Furthermore, the fact that only the proband presented this association—and not among the rest of the relatives—is an example of the phenotypic variability of the disease.
Previously, the c.14465G > A, p.Arg4822His variant detected has only been reported in one individual in a cohort of patients with CPVT [1, 5] without providing any clinical or co-segregation information. The case we present supports the pathogenicity of the genetic variant in RYR2 according to the American College of Medical Genetics (ACMG) criteria since it has not been detected in large populations of healthy controls, the patients present a characteristic phenotype of CPVT and additionally, a co-segregation between the members of the family is also observed.
Several attempts have been made in this field to establish a relationship between the structural alteration and the most malignant phenotypes, thereby observing that variants affecting the C-terminal region and other key areas to regulate channel activation cause the most serious conditions [3]. In our case, the variant c.14465G > A, p.Arg4822His does not cover the regions involved in severe phenotypes. It is a conservative amino acid substitution, as it does not affect the secondary structure of the protein given that the residues share similar physicochemical properties [6, 7]. Both these reasons could explain the good prognosis of the family considered in our study.
Conclusions
We described, for the first time, the clinical characteristics and co-segregation of a family diagnosed with CPVT secondary to a little-known genetic variant of the RYR2 gene. In our case, the variant suggests association with a very good prognosis and diagnosis at late adulthood and exhibits an excellent response to treatment with beta-blockers.
Availability of data and materials
The authors declare that the data supporting the findings of this study are available within this article.
Abbreviations
- CPVT:
-
Catecholaminergic polymorphic ventricular tachycardia
- VT:
-
Ventricular tachycardia
- ECG:
-
Electrocardiogram
- RyR2:
-
Ryanodine receptor 2
- PVCs:
-
Premature ventricular complexes
- NGS:
-
Next-Generation Sequencing
- ACMG:
-
American College of Medical Genetics
References
Medeiros-Domingo A, Bhuiyan ZA, Tester DJ, et al. The RYR2-encoded ryanodine receptor/calcium release channel in patients diagnosed previously with either catecholaminergic polymorphic ventricular tachycardia or genotype negative, exercise-induced long QT syndrome. A comprehensive open reading frame mutational analysis. J Am Coll Cardiol. 2009;54(22):2065–74. https://doi.org/10.1016/j.jacc.2009.08.022.
Sy RW, Gollob MH, Klein GJ, et al. Arrhythmia characterization and long-term outcomes in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2011;8(6):864–71. https://doi.org/10.1016/j.hrthm.2011.01.048.
Roston TM, Yuchi Z, Kannankeril PJ, et al. The clinical and genetic spectrum of catecholaminergic polymorphic ventricular tachycardia: findings from an international multicentre registry. Europace. 2018;20(3):541–7. https://doi.org/10.1016/j.hrthm.2011.01.048.
Brunetti ND, Sai R, Gaglione A, et al. Catecholaminergic polymorphic ventricular tachycardia associated with sinus node dysfunction and junctional rhythm: case report and literature review. J Electrocardiol. 2016;49(6):940–3. https://doi.org/10.1016/j.jelectrocard.2016.07.024.
Walsh R, Peters NS, Cook SA, Ware JS. Paralogue annotation identifies novel pathogenic variants in patients with Brugada syndrome and catecholaminergic polymorphic ventricular tachycardia. J Med Genet. 2014;51(1):35–44. https://doi.org/10.1136/jmedgenet-2013-1019.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24. https://doi.org/10.1038/gim.2015.30.
National Center for Biotechnology Information. ClinVar [VCV000201358.2] https://www.ncbi.nlm.nih.gov/clinvar/variation/201358/evidence/
Funding
This research received no grant from any funding agency in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
LG, TB, and AG contributed to the study conception and design. LG, TB, and AG contributed to the acquisition of data. LG, TB, and AG had the lead in preparing the manuscript. JI, AI, MM, and JM critically revised the manuscript. TB and AG were involved in the clinical management of the patient. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Written informed consent for the publication of this case report and any accompanying images was obtained from the patient. All authors have agreed to the content of this manuscript and agree to its submission to the journal.
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
García-Cano, L., Brouzet, T.A., García-Fernández, A. et al. Diagnosis of catecholaminergic polymorphic ventricular tachycardia during late adulthood due to a rare genetic variant in RYR2: a case report. Int J Arrhythm 22, 13 (2021). https://doi.org/10.1186/s42444-021-00043-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42444-021-00043-x