
Abstract
Introduction
The deficiency of ADA2 (DADA2) is a rare autoinflammatory disease provoked by mutations in the ADA2 gene inherited in a recessive fashion. Up to this moment there is no consensus for the treatment of DADA2 and anti-TNF is the therapy of choice for chronic management whereas bone marrow transplantation is considered for refractory or severe phenotypes. Data from Brazil is scarce and this multicentric study reports 18 patients with DADA2 from Brazil.
Patients and methods
This is a multicentric study proposed by the Center for Rare and Immunological Disorders of the Hospital 9 de Julho - DASA, São Paulo - Brazil. Patients of any age with a confirmed diagnosis of DADA2 were eligible for this project and data on clinical, laboratory, genetics and treatment were collected.
Results
Eighteen patients from 10 different centers are reported here. All patients had disease onset at the pediatric age (median of 5 years) and most of them from the state of São Paulo. Vasculopathy with recurrent stroke was the most common phenotype but atypical phenotypes compatible with ALPS-like and Common Variable Immunodeficiency (CVID) was also found. All patients carried pathogenic mutations in the ADA2 gene. Acute management of vasculitis was not satisfactory with steroids in many patients and all those who used anti-TNF had favorable responses.
Conclusion
The low number of patients diagnosed with DADA2 in Brazil reinforces the need for disease awareness for this condition. Moreover, the absence of guidelines for diagnosis and management is also necessary (t).
Similar content being viewed by others


Introduction
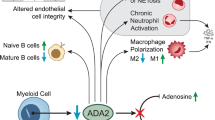
The Deficiency of deaminase 2 (DADA2) is an inborn error of immunity, a rare autoinflammatory disease (MIM #615,688) first described in 2014 [1]. It is a recessive disorder, caused by biallelic mutations in the ADA2 gene (previously named CERC1 - cat eye syndrome chromosome region, candidate 1). ADA (adenosine deaminase) is a key enzyme of the purine metabolism pathway responsible for immunologic functions by catalyzing the adenosine to inosine which is a key immunosuppressive signal [2]. On one hand, ADA1 has a higher affinity to adenosine, is more expressed as an intracellular protein and its deficiency leads to the SCID (severe combined immunodeficiency) T-/B-/NK- phenotype. On the other hand, ADA2 is a plasma secreted enzyme that is important to co-regulate immune cells, especially in the differentiation of macrophages into monocytes, the major source of ADA2 [3].
The immune-phato-physiology of DADA2 remains elusive and this topic was recently approached by Ehler and Meyts in an insightful editorial of the role of ADA2 in DADA2 deficiency [4]. The absence of ADA2, in co-culture cells experiments, demonstrated to cause a disturbance in endothelial cells leading to vasculopathy. Thereafter zebrafish ADA2 knockout models exhibited intracranial hemorrhagic stroke that ameliorated after ADA2 injection, a proven model that ADA2 is essential for the endothelial homeostasis [1]. ADA2 also is related to increased NET-formation associated with TNF-alpha secretion [5]. Recently, positive levels of interferon stimulated genes (ISGs) were found positive in patients with DADA2 which surrogated the role of the interferons in ADA2 biology [6]. IFN-B seems to induce skewing of macrophages into proinflammatory M1 cells leading to TNF-alpha secretion but is more a consequence than a cause of immunologic phenomenon observed in DADA2 [4]. Finally, Watanabe et al. recently spotted the light back to the monocytes as they demonstrated an enrichment of nonclassical monocytes in peripheral blood of DADA2 patients aligned with high levels of TNF-alpha, according to previous demonstrated studies [7].
From the clinical point of view, many points still require discussion in DADA2 diagnosis and management, as there is no consensus for this disorder. The initial descriptions of DADA2 reported this disorder solely as a systemic associated vasculopathy associated disorder [1]. The first phenotypes were mainly characterized by recurrent fever associated with cutaneous vasculitis (e.g.: polyarteritis nodosa) and recurrent episodes of stroke. Over the years an expansion on the phenotypes related to DADA2 have been reported with findings consistent with pure cell aplasia, hemolytic anemia, hypogammaglobulinemia. childhood lymphoma and more recently associated with the ALPS-like phenotype [8,9,10,11]. Although the final diagnosis of DADA2 is genetic, low or absent levels of ADA2 in peripheral blood can fastly assist the diagnosis in cases of emergency and support the final diagnosis in situations of variants of uncertain significance (VUS) or findings of single mutations [12]. Therefore, the lack of sources may turn the diagnosis of DADA2 even more difficult in middle-incoming countries.
DADA2 has been reported worldwide and data from Brazil are scarce and results from independent case reports [13,14,15]. In this paper we report clinic, genetic and therapeutic data of a multicentric study of Brazilian patients with DADA2.
Patients and methods
We conducted a retrospective multicenter study in Brazil from January/2019-december/2022. Reference centers for inborn errors of immunity (IEI) from Brazil were invited to participate throughout the network of the Center for Rare and Immunological Disorders of the Hospital 9 de Julho (DORAID - H9J/DASA and the Brazilian Association of Autoinflammatory Diseases (ANDAI). After ethical approval (Hospital 9 de Julho - Rede DASA; CAAE: 48301321.9.1001.54.55) and patients consent to participate, records were revised and data on clinical manifestations, laboratory workup, genetic analysis, treatments were collected. Response to treatment was considered complete when clinical and laboratory remission could be achieved; partial when clinical or laboratory remission could not be achieved; and absent when nor clinical or laboratory remission could be achieved. Clinical remission was defined as the absence of fever and control of the skin manifestation and/or of signs of lymphoproliferation. Laboratory remission was considered after normalization of CRP levels in the local laboratory. Statistical analysis was carried out using Prism 8.0.
Results
10 centers from all over Brazil agreed to participate in this multicentric study. We found 18 patients with a female predominance (67%, n = 12), Table 1. Most were originally from the southeast of Brazil (55%; n = 10) and south (27%; n = 5) of Brazil (Table 1; Fig. 1). The median age of initial symptoms was 5 years with a median delay to diagnosis of 17 years (min 2,5; max 43). Broad spectrum of syndromic presentation could be recognized as presented in Table 1, and the predominant phenotype was the classical polyarteritis nodosa and recurrent stroke, found in half of the patients. P2 presented with a severe pancytopenia and signs of hemolytic anemia. P7 was diagnosed with Hodgkin lymphoma at the age of 4 years, remained asymptomatic for 5 years and then started with recurrent fever, lymphoproliferation and cytopenia, compatible with ALPS-like phenotype. P8 presented with recurrent fever associated with diffuse lymphoproliferation and anemia that was initially labeled as ALPS-like phenotype. And finally, P16 and P18 presented as hypogammaglobulinemia, and recurrent sinusitis initially characterized as common variable immunodeficiency (CVID). P10-12 received the syndromic diagnosis of antiphospholipid syndrome (APS).

Geographical distribution in Brazil per state of DADA2 and centers with DADA2 experience found in this study. On the right side, the acronym used for the Brazilian states
Fever could not be observed just in 5 patients, mainly those with atypical phenotypes (P10,11, 13, 16 and P18). Most of the patients had a continuous disease pattern (73%) rather than periodic. When periodic, fever episodes were associated with systemic inflammation (high levels of CRP) with skin manifestation or stroke, lasting 4,5 days on average and this pattern could be observed in P1, P2, P3, P4 and P17. Mucocutaneous, musculoskeletal and neurological signs were the core characteristics of DADA2 in Brazil. Livedo reticularis was found in more than half of the patients. Overall, arthralgia was found in 45% (n = 8) and myalgia in 33% (n = 6) whereas both signs could be observed in 16% (n = 2; P6, P7, P14 and P17). Severe and disabling abdominal pain was found in 45% of the patients and two patients had detectable gastrointestinal lesions on colonoscopy with pathologic findings compatible with nonspecific ulcers and colitis in P7 and with intestinal necrosis due to small vessels vasculitis in P6. Signs of diffuse lymphoproliferation were observed in 34% (n = 6). Hepato and splenomegaly was found in 23% (P1, P2, P8 and P18) whereas P5 and P6 presented only splenomegaly. All clinical data are reported in Table 2. Additionally persistent neutropenia was a constant finding in P4. Hypogammaglobulinemia was found in P1, P2, P7, P16 and P18.
All patients harbor pathogenic or likely pathogenic mutations in the ADA2 gene, as summarized in Table 1. There was no genotype and phenotype correlation, no predominant/founder mutation and no predominant exonic locations of the mutations. Most of the patients, 60% (n = 10) carried homozygous mutations, and the Y453C was the most common variant (n = 7), followed by c.973-2 A (n = 6) splice acceptor. Two patients harbored novel mutations, P7 and P18, with reduced expression of ADA2 in western-blot assay (data not shown). P7 also was found to have positive interferon ISGs (Interferon score of 8,22 – local laboratory refence of: [n = 29] mean 240,6; Min 0,9; Max 3264,8; SD 628,86). Familial clusters could be observed in two families: Family X - P10, P11, P12; and Family XI - P13 and P14 (Supplementary File 1).
At least 62% (n = 11) patients used steroids (prednisolone mainly) during flares and a curiously unsatisfactory response could be observed in 6 patients (Table 3). Cyclophosphamide was also not effective in 3 patients as an induction therapy. P2 and P5 did not experience complete control of disease under chronic immunoglobulin replacement (dose of 500 mg/kg every 28 days). Unsatisfactory responses to mycophenolate, azathioprine and sirolimus could be observed in all those who used these drugs. Mycophenolate had an accumulated benefit to P2 who relapsed from hemolytic anemia after the second shot of etanercept. Different anti-TNF choices could be observed all over Brazil and inadequate response was observed just in one patient under infliximab (P17) over 10 months of follow-up. P5, P6, P7 and P9 achieved complete clinical control after adalimumab was initiated (median follow-up of 20 months; min 12; max 24 months). P1, P2, P3, P4, P10, P11, P12, P13 and P15 all received etanercept with complete control of disease after a median follow up of 56 months; min 36; max 72 months (Table 3). Certolizumab was used only in P14 with complete control of the disease over a follow-up of 6 months. We have not identified any DADA2 patient transplanted in Brazil and P18 was sent to bone marrow transplant evaluation at the last visit.
Intriguing, identifiable triggers for the disease could be noted in 41% (n = 5) of the patients, being cold exposure in 25% (P1, P10, P11 and P12) and infection was also reported just by P1. Disabilities related to the disease could be found in 50% (n = 9), Table 2. Up to the last follow-up two patients died (2/18; 12%), P7 (disseminated staphylococcal infection) and P8 (severe hemorrhagic stroke).
Discussion
In this multicentric study we report 18 patients with DADA2 from Brazil, 5 of them already reported [13,14,15] (Fig. 1). The exact frequency of DADA2 in the world is not yet established but it is estimated to affect as high as 4:100.000 individuals [16]. Considering the Brazilian population at the last census in 2020 of 212 million, the low number of individuals identified and the limited number of centers with patients emphasizes the urgent need of disease awareness. Therefore, as DADA2 can be found in many clinical scenarios and medical specialties we also present here a geographical distribution of centers where patients with DADA2 are followed to redirect any patient or physician in need (Fig. 1).
The absence of diagnostic criteria to DADA2 coupled with limited access to functional assays and genetic sequencing in middle income countries may also be taken into account when talking about the low number of patients herein identified. Another important consideration for the diagnosis of DADA2 is the pleiotropic manifestation of the disease [8]. As we found, DADA2 can be recognized among at least three main syndromic phenotypes: vasculitis (polyarteritis nodosa), recurrent stroke (hemorrhagic or ischemic) and the ALPS-like phenotype (with or without hypogammaglobulinemia). However, the high rate of disabilities found when the diagnosis is made at that level suggests that actions should be paid to a step before, but how? As we demonstrated, there is a high variability of signs and symptoms among all syndromic presentations, therefore, early clinical diagnosis is tricky. In conclusion, the general use of enzymatic measurement and the application of general genetic sequencing may be the answer. Another question still not answered that may implicate in phenotype variability is the exposome consequence in those patients affected, and so different phenotypes within the same family with epigenetics variations may give us a clue.
The enzymatic activity of ADA2 may be a strong screening method for DADA2, however, many questions are still to be answered [17, 18]. As previously said, ADA 1 and 2 differ in expression, cellular localization, catalytic properties and are associated with different phenotypes in the IEI. The dynamics of ADA2 levels in the general and among different populations are not yet known. So far, it seems to diminish with aging and high levels can be found in many other situations, including other disorders of systemic inflammation and cancer. Yet, it is still unclear if low/undetectable levels is an exclusive fund of DADA2. For diagnosis purposes in DADA2 many different protocols for ADA2 levels detection have been published, and cost effectiveness of different ones has not yet been evaluated. Using a sandwich ELISA design, very low levels of ADA2 were found in serum of affected patients (children and adults), while normal levels were found in healthy controls, the same group observed medium levels of ADA2 (higher than in the affected) in carriers of ADA2 mutations [19,20,21,22,23]. Another interesting finding herein reported is the presence of low interferon score in one DADA2 patient, as previously reported [6]. If this finding could be useful for diagnosis, to identify peculiar phenotypes, to guide any specific therapy or if it is of any relevance for physiopathology, it is also an unanswered question.
The genetics of DADA2 is the hallmark for the diagnosis and some interesting findings have been already reported. Lee et al. found that residual (~ 3%) of ADA2 enzyme correlates with the classical phenotypes of vasculitis and missense mutations, whereas very low (undetectable) levels was found in patients with a more prominent hematological phenotype (bone marrow failure) [24]. Although we have not performed ADA2 levels in all our patients, in our cohort we can observe that the presence of deletions/insertions was found only in more hemato/immunological phenotypes that support the presence of genotype and phenotype correlations in DADA2 patients. The Brazilian population is of great admixture and variety in genetic findings is expected, and so our findings of a novel mutation affecting the ADA2 gene here reported expands the genotype findings of DADA2. Indeed, as we found, the splice acceptor mutation (c.973-2 A − 33% in this study) is supposed to be a common finding in populations of genetic admixture, such as Brazil. Furthermore, we also observed that the Y453C is also common among DADA2 patients in Brazil (~ 40% in this study). In the ABRAOM, repository for genomic variants for the Brazilian population, both predominant variants found are also rare and cannot be found in homozygous state (the c.973-2 A has an estimated frequency of 0,001708 and no homozygotes can be observed and the Y453C of 0,000584 also without any homozygote) [25].
Finally, regarding the treatment of ADA2, general acute management of vasculitis seems not to be efficient in all patients as we reported in Table 3. As observed, not all patients had satisfactory responses to high doses of steroids or cyclophosphamide, considering that this is a retrospective study. For acute management, previous reports rationally suggested the use of plasma infusion as a replacement for ADA2 in deficient patients, but this has no longer been evaluated [26]. Also, as we observed, chronic infusions of IVIG seem to not prevent thrombotic events, as we observed in patients previously labeled as CVID under chronic IVIG replacement [18]. No other immunosuppressive therapy appears to be effective in controlling DADA2. Anti-TNF drugs revolutionized the treatment of DADA2, and variable responses to different anti-TNF have been reported. Here we have observed that only infliximab was not effective in only one patient in a short period of follow-up [27]. Finally, hematopoietic stem cell transplantation is the therapy of choice for refractory/recalcitrant diseases and was found to be curative for many individuals, but the risk of complication must be considered during the procedure and chronic graft versus host disease is also a feared complication [28]. Gene therapy may be a great option for DADA2 patients in the near future as recently demonstrated in preclinical studies [29].
Conclusion: This is the first multicenter study of DADA2 in Brazil that reinforces the need for disease awareness in the country due to the low number of individuals identified. We also demonstrate the same phenotypic and genotypic variability of DADA2 in Brazilian as previously found in other countries. Although we have observed the same response to anti-TNF drugs in Brazilian DADA2 patients a consensus is necessary to answer many other clinical questions.
Data availability
The authors confirm that the data supporting the findings of this study are available within the corresponding author when requested.
References
Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Zavialov AV, Stone DL, Chae JJ, Rosenzweig SD, Bishop K, Barron KS, Kuehn HS, Hoffmann P, Negro A, Tsai WL, Cowen EW, Pei W, Milner JD, Silvin C, Heller T, Chin DT, Patronas NJ, Barber JS, Lee CC, Wood GM, Ling A, Kelly SJ, Kleiner DE, Mullikin JC, Ganson NJ, Kong HH, Hambleton S, Candotti F, Quezado MM, Calvo KR, Alao H, Barham BK, Jones A, Meschia JF, Worrall BB, Kasner SE, Rich SS, Goldbach-Mansky R, Abinun M, Chalom E, Gotte AC, Punaro M, Pascual V, Verbsky JW, Torgerson TR, Singer NG, Gershon TR, Ozen S, Karadag O, Fleisher TA, Remmers EF, Burgess SM, Moir SL, Gadina M, Sood R, Hershfield MS, Boehm M, Kastner DL, Aksentijevich I. Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med. 2014 Mar;370(6):911–20. https://doi.org/10.1056/NEJMoa1307361. Epub 2014 Feb 19. PMID: 24552284; PMCID: PMC4193683.
Gao ZW, Yang L, Liu C, Wang X, Guo WT, Zhang HZ, Dong K. Distinct roles of Adenosine Deaminase Isoenzymes ADA1 and ADA2: a Pan-Cancer Analysis. Front Immunol. 2022 May;18:13:903461. https://doi.org/10.3389/fimmu.2022.903461. PMID: 35663977; PMCID: PMC9157497.
Kaljas Y, Liu C, Skaldin M, Wu C, Zhou Q, Lu Y, Aksentijevich I, Zavialov AV. Human adenosine deaminases ADA1 and ADA2 bind to different subsets of immune cells. Cell Mol Life Sci. 2017 Feb;74(3):555–70. Epub 2016 Sep 23. PMID: 27663683.
Ehlers L, Meyts I. What a difference ADA2 makes: insights into the pathophysiology of ADA2 deficiency from single-cell RNA sequencing of monocytes. J Leukoc Biol. 2021 Sep;110(3):405–7. https://doi.org/10.1002/JLB.5CE0421-186R. Epub 2021 Jun 16. PMID: 34137068.
Carmona-Rivera C, Khaznadar SS, Shwin KW, Irizarry-Caro JA, O’Neil LJ, Liu Y, Jacobson KA, Ombrello AK, Stone DL, Tsai WL, Kastner DL, Aksentijevich I, Kaplan MJ, Grayson PC. Deficiency of adenosine deaminase 2 triggers adenosine-mediated NETosis and TNF production in patients with DADA2. Blood 2019 Jul 25;134(4):395–406. doi: https://doi.org/10.1182/blood.2018892752. Epub 2019 Apr 23. PMID: 31015188; PMCID: PMC6659253.
Insalaco A, Moneta GM, Pardeo M, Caiello I, Messia V, Bracaglia C, Passarelli C, De Benedetti F. Variable clinical phenotypes and relation of Interferon signature with Disease Activity in ADA2 Deficiency. J Rheumatol. 2019 May;46(5):523–6. https://doi.org/10.3899/jrheum.180045. Epub 2019 Jan 15. PMID: 30647181.
Watanabe N, Gao S, Wu Z, Batchu S, Kajigaya S, Diamond C, Alemu L, Raffo DQ, Hoffmann P, Stone D, Ombrello AK, Young NS. Analysis of deficiency of adenosine deaminase 2 pathogenesis based on single-cell RNA sequencing of monocytes. J Leukoc Biol 2021 Sep;110(3):409–24. doi: https://doi.org/10.1002/JLB.3HI0220-119RR. Epub 2021 May 14. PMID: 33988272.
Barron KS, Aksentijevich I, Deuitch NT, Stone DL, Hoffmann P, Videgar-Laird R, Soldatos A, Bergerson J, Toro C, Cudrici C, Nehrebecky M, Romeo T, Jones A, Boehm M, Kanakry JA, Dimitrova D, Calvo KR, Alao H, Kapuria D, Ben-Yakov G, Pichard DC, Hathaway L, Brofferio A, McRae E, Moura NS, Schnappauf O, Rosenzweig S, Heller T, Cowen EW, Kastner DL, Ombrello AK. The Spectrum of the Deficiency of Adenosine Deaminase 2: An Observational Analysis of a 60 Patient Cohort. Front Immunol 2022 Jan 10;12:811473. doi: https://doi.org/10.3389/fimmu.2021.811473. PMID: 35095905; PMCID: PMC8790931.
Pilania RK, Banday AZ, Sharma S, Kumrah R, Joshi V, Loganathan S, Dhaliwal M, Jindal AK, Vignesh P, Suri D, Rawat A, Singh S. Deficiency of Human Adenosine Deaminase Type 2 - A Diagnostic Conundrum for the Hematologist. Front Immunol 2022 May 3;13:869570. doi: https://doi.org/10.3389/fimmu.2022.869570. PMID: 35592317; PMCID: PMC9110783.
Alsultan A, Basher E, Alqanatish J, Mohammed R, Alfadhel M. Deficiency of ADA2 mimicking autoimmune lymphoproliferative syndrome in the absence of livedo reticularis and vasculitis. Pediatr Blood Cancer. 2018 Apr;65(4). https://doi.org/10.1002/pbc.26912. Epub 2017 Dec 22. PMID: 29271561.
Alabbas F, Elyamany G, Alsharif O, Hershfield M, Meyts I. Childhood Hodgkin Lymphoma: think DADA2. J Clin Immunol. 2019 Jan;39(1):26–9. https://doi.org/10.1007/s10875-019-0590-7. Epub 2019 Jan 14. PMID: 30644014.
Aksentijevich I, Sampaio Moura N, Barron K. Adenosine Deaminase 2 Deficiency. 2019 Aug 8. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2022. PMID: 31393689.
Ferriani MPL, Valera ET, de Sousa GR, Sandrin-Garcia P, de Moura RR, Hershfield MS, de Carvalho LM. ADA2 deficiency (DADA2) associated with Evans syndrome and a severe ADA2 genotype. Rheumatology (Oxford). 2021 Jul 1;60(7):e237-e239. doi: https://doi.org/10.1093/rheumatology/keab011. PMID: 33493352.
Chong-Neto HJ, Segundo GRS, Bandeira M, Chong-Silva DC, Rosário CS, Riedi CA, Hershfield MS, Ochs H, Torgerson T, Rosário NA. Homozygous splice ADA2 gene mutation causing ADA-2 Deficiency. J Clin Immunol. 2019 Nov;39(8):842–5. https://doi.org/10.1007/s10875-019-00697-2. Epub 2019 Oct 15. PMID: 31617030.
Poswar Fde O, da Fonseca RM, de Albuquerque LC, Zhou Q, Jardim LB, Monte TL, Aksentijevich I, Saute JA. Adenosine deaminase 2 deficiency presenting as spastic paraplegia and systemic vasculitis. J Neurol. 2016 Apr;263(4):818–20. Epub 2016 Feb 25. PMID: 26914925.
Jee H, Huang Z, Baxter S, Huang Y, Taylor ML, Henderson LA, Rosenzweig S, Sharma A, Chambers EP, Hershfield MS, Zhou Q, Dedeoglu F, Aksentijevich I, Nigrovic PA, O’Donnell-Luria A, Lee PY. Comprehensive analysis of ADA2 genetic variants and estimation of carrier frequency driven by a function-based approach. J Allergy Clin Immunol. 2022 Jan;149(1):379–87. Epub 2021 May 15. PMID: 34004258; PMCID: PMC8591146.
Abbasi A, Batllori M, Gil-Sáez FJ, Rodríguez-Pintó I, Antón López J, Iglesias Jímenez E. Importance of the determination of enzymatic activity in the diagnosis of deficiency of adenosine deaminase 2 (DADA2). Med Clin (Barc). 2022 Sep 23;159(6):283–286. English, Spanish. doi: https://doi.org/10.1016/j.medcli.2021.12.020. Epub 2022 Feb 28. PMID: 35241284.
Schepp J, Proietti M, Frede N, Buchta M, Hübscher K, Rojas Restrepo J, Goldacker S, Warnatz K, Pachlopnik Schmid J, Duppenthaler A, Lougaris V, Uriarte I, Kelly S, Hershfield M, Grimbacher B. Screening of 181 Patients With Antibody Deficiency for Deficiency of Adenosine Deaminase 2 Sheds New Light on the Disease in Adulthood. Arthritis Rheumatol. 2017 Aug;69(8):1689–1700. doi: https://doi.org/10.1002/art.40147. Epub 2017 Jul 5. PMID: 28493328.
Luo W, Dong L, Chen F, Lei W, He L, Zhou Q, Lamy T, Zavialov AV. ELISA based assays to measure adenosine deaminases concentration in serum and saliva for the diagnosis of ADA2 deficiency and cancer. Front Immunol 2022 Jul 28;13:928438. doi: https://doi.org/10.3389/fimmu.2022.928438. PMID: 35967411; PMCID: PMC9366848.
Ito M, Nihira H, Izawa K, Yasumi T, Nishikomori R, Iwaki-Egawa S. Enzyme activity in dried blood spot as a diagnostic tool for adenosine deaminase 2 deficiency. Anal Biochem. 2021 Sep 1;628:114292. doi: https://doi.org/10.1016/j.ab.2021.114292. Epub 2021 Jun 24. PMID: 34171384.
Poursharifi P, Saghiri R, Ebrahimi-Rad M, Nazem H, Pourpak Z, Moin M, Shams S. Adenosine deaminase in patients with primary immunodeficiency syndromes: the analysis of serum ADA1 and ADA2 activities. Clin Biochem. 2009 Sep;42(13–14):1438–43. https://doi.org/10.1016/j.clinbiochem.2008.10.019. Epub 2008 Nov 8. PMID: 19026999.
Franco-Martínez L, Tecles F, Torres-Cantero A, Bernal E, San Lázaro I, Alcaraz MJ, Vicente-Romero MR, Lamy E, Sánchez-Resalt C, Rubio CP, Tvarijonaviciute A, Martínez-Subiela S, Cerón JJ. Analytical validation of an automated assay for the measurement of adenosine deaminase (ADA) and its isoenzymes in saliva and a pilot evaluation of their changes in patients with SARS-CoV-2 infection. Clin Chem Lab Med. 2021 Apr 28;59(9):1592–1599. doi: https://doi.org/10.1515/cclm-2021-0324. PMID: 33908223.
Bowers SM, Gibson KM, Cabral DA, Brown KL. Adenosine deaminase 2 activity negatively correlates with age during childhood. Pediatr Rheumatol Online J 2020 Jul 10;18(1):54. doi: https://doi.org/10.1186/s12969-020-00446-5. PMID: 32650798; PMCID: PMC7350767.
Lee PY, Kellner ES, Huang Y, Furutani E, Huang Z, Bainter W, Alosaimi MF, Stafstrom K, Platt CD, Stauber T, Raz S, Tirosh I, Weiss A, Jordan MB, Krupski C, Eleftheriou D, Brogan P, Sobh A, Baz Z, Lefranc G, Irani C, Kilic SS, El-Owaidy R, Lokeshwar MR, Pimpale P, Khubchandani R, Chambers EP, Chou J, Geha RS, Nigrovic PA, Zhou Q. Genotype and functional correlates of disease phenotype in deficiency of adenosine deaminase 2 (DADA2). J Allergy Clin Immunol. 2020 Jun;145(6):1664–1672e10. Epub 2020 Jan 13. PMID: 31945408; PMCID: PMC7282972.
Naslavsky MS, Scliar MO, Yamamoto GL, Wang JYT, Zverinova S, Karp T, Nunes K, Ceroni JRM, de Carvalho DL, da Silva Simões CE, Bozoklian D, Nonaka R, Dos Santos Brito Silva N, da Silva Souza A, de Souza Andrade H, Passos MRS, Castro CFB, Mendes-Junior CT, Mercuri RLV, Miller TLA, Buzzo JL, Rego FO, Araújo NM, Magalhães WCS, Mingroni-Netto RC, Borda V, Guio H, Rojas CP, Sanchez C, Caceres O, Dean M, Barreto ML, Lima-Costa MF, Horta BL, Tarazona-Santos E, Meyer D, Galante PAF, Guryev V, Castelli EC, Duarte YAO, Passos-Bueno MR, Zatz M. Whole-genome sequencing of 1,171 elderly admixed individuals from São Paulo, Brazil. Nat Commun. 2022 Mar 4;13(1):1004. doi: https://doi.org/10.1038/s41467-022-28648-3. Erratum in: Nat Commun. 2022 Mar 30;13(1):1831. PMID: 35246524; PMCID: PMC8897431.
Ombrello AK, Qin J, Hoffmann PM, Kumar P, Stone D, Jones A, Romeo T, Barham B, Pinto-Patarroyo G, Toro C, Soldatos A, Zhou Q, Deuitch N, Aksentijevich I, Sheldon SL, Kelly S, Man A, Barron K, Hershfield M, Flegel WA, Kastner DL. Treatment Strategies for Deficiency of Adenosine Deaminase 2. N Engl J Med. 2019 Apr 18;380(16):1582–1584. doi: https://doi.org/10.1056/NEJMc1801927. PMID: 30995379; PMCID: PMC7372950.
Meyts I, Aksentijevich I. Deficiency of Adenosine Deaminase 2 (DADA2): updates on the phenotype, Genetics, Pathogenesis, and treatment. J Clin Immunol. 2018 Jul;38(5):569–78. https://doi.org/10.1007/s10875-018-0525-8. Epub 2018 Jun 27. PMID: 29951947; PMCID: PMC6061100.
Hashem H, Bucciol G, Ozen S, Unal S, Bozkaya IO, Akarsu N, Taskinen M, Koskenvuo M, Saarela J, Dimitrova D, Hickstein DD, Hsu AP, Holland SM, Krance R, Sasa G, Kumar AR, Müller I, de Sousa MA, Delafontaine S, Moens L, Babor F, Barzaghi F, Cicalese MP, Bredius R, van Montfrans J, Baretta V, Cesaro S, Stepensky P, Benedicte N, Moshous D, Le Guenno G, Boutboul D, Dalal J, Brooks JP, Dokmeci E, Dara J, Lucas CL, Hambleton S, Wilson K, Jolles S, Koc Y, Güngör T, Schnider C, Candotti F, Steinmann S, Schulz A, Chambers C, Hershfield M, Ombrello A, Kanakry JA, Meyts I. Hematopoietic Cell Transplantation Cures Adenosine Deaminase 2 Deficiency: Report on 30 Patients. J Clin Immunol. 2021 Oct;41(7):1633–1647. doi: https://doi.org/10.1007/s10875-021-01098-0. Epub 2021 Jul 29. Erratum in: J Clin Immunol. 2022 Oct;42(7):1580–1581. PMID: 34324127; PMCID: PMC8452581.
Hong Y, Casimir M, Houghton BC, Zhang F, Jensen B, Omoyinmi E, Torrance R, Papadopoulou C, Cummins M, Roderick M, Thrasher AJ, Brogan PA, Eleftheriou D. Lentiviral Mediated ADA2 Gene Transfer Corrects the Defects Associated With Deficiency of Adenosine Deaminase Type 2. Front Immunol 2022 Apr 22;13:852830. doi: https://doi.org/10.3389/fimmu.2022.852830. PMID: 35529868; PMCID: PMC9073084.
Acknowledgements
The authors would like to thanks the patients and their families for their participation in this study.
Funding
This paper was supported by the Center for Rare and Immunological Disorders - Hospital 9 de Julho/DASA.
Author information
Authors and Affiliations
Contributions
All authors contributed equally to the construction of this manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This project was conducted under the (DADA2 Project - Hospital 9 de Julho - DASA; CAAE: 48301321.9.1001.54.55) and (DORAID - Doenças Raras e da Imunidade Project - Hospital 9 de Julho - DASA; CAAE: 58091122.4.0000.5455).
Consent for publication
All centers and patients have consented for publication of this paper.
Competing interests
The authors declare no competing interests regarding the publication of this paper.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Melo, A., de Carvalho, L.M., Ferriani, V.P.L. et al. A brazilian nationwide multicenter study on deficiency of deaminase-2 (DADA2). Adv Rheumatol 63, 23 (2023). https://doi.org/10.1186/s42358-023-00303-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42358-023-00303-5