
Abstract
Background
Dengue virus (DENV) infection is spreading rapidly, especially in the subtropical and tropical regions, placing a huge percentage of the global population at risk and causing repeated outbreaks. DENV protease inhibition has been suggested as a viable therapeutic strategy. Using a computer-aided design approach and the structure-based drug design approach, ten 1, 2, 4-oxadiazole derivatives were designed based on the lead template (34) from our prior study. The design involved the substitution at the phenyl pharmacophore of the lead with methylamine, hydroxyl, and methoxy groups. To compare the anti-DENV efficacy of the optimized designed compounds to the template and other DENV referenced inhibitors targeting the NS-5 protease (PDB ID: 5K5M), they were docked with the DENV NS-5 protease. In silico, ADME characteristics and drug-likeness were also assessed for the compounds.
Results
The molecular docking scores of the designed 1, 2, 4-oxadiazole derivatives varied from − 19.091 to − 29.61 kcal/mol, with excellent hydrogen bond energies in the range of − 3.402 to − 9.0128 kcal/mol, compared to the lead with a score of − 19.10 kcal/mol, and the hydrogen bond energy is − 3.10 kcal/mol, both of which are lower than those of the proposed compounds. Ferentinide, S-adenosyl-l-homocysteine, and Ribavirin were found to have lower binding scores of − 15.8137, − 16.5362, and − 12.446 kcal/mol, respectively, with hydrogen bond energies of − 4.2659, − 10.4372, and − 7.2995 kcal/mol. The developed compounds all followed Lipinski's criteria, meaning they were highly bioavailable, had no potential carcinogenic or mutagenic properties, and posed no concern of cardiovascular toxicity based on the ADMET profile.
Conclusion
The proposed oxadiazole derivative interacted better with DENV protease (NS-5) than the lead inhibitor as well as the conventional inhibitors. Compounds 34a and 34b had the best ligand-protease interaction and gave the lowest free energy at − 26.54 and − 29.612 kcal/mol, respectively. Hence, they could be suggested as potential therapeutic candidates to inhibit NS-5 RdRp protease. This study has revealed the anti-DENV action of the designed compounds, indicating that synthesis and in vivo studies into their efficacy and mechanism are warranted.
Similar content being viewed by others

Background
The DENV which belongs to the Flavivirus family vectored through female mosquitoes is the main cause of dengue fever disease which has spread to various regions of the world at an epidemic rate especially in the tropical and sub-tropical region, thereby posing great danger to the vast population of the world in recent time (Bhatt et al. 2013). The health risks posed by DENV infection include slight flu-like signs, to the further severe hemorrhagic fever or shock syndrome that could lead to death if unattended to (Megawati et al. 2017; Wilder-Smith et al. 2017; Batool et al. 2021).
There are about four known serotypes of the DENV (1, 2, 3, and 4), in which any could cause the infection but infection by one of the serotypes could offer immunity against further infection but not against other serotypes; infected individuals are susceptible to further infection due to antibody-dependent disease enhancement. Among the seven nonstructural (NS-1, NS-2a, NS-2b, NS-3, NS-4a, NS-4b, and NS-5) proteins of the DENV, the NS-5 is the most conserved among all the serotypes that having an essential part in the DENV duplication thereby serving as the target of interest for the treatment of the DENV infection (Anusuya and Gromiha 2019).
The alarming rate of DENV infection coupled with undesirable health impacts on children and previously infected persons as well as the unavailability of approved drugs demand urgent attention towards the discovery of a more potent therapeutic agent to fight the scourge of the infection.
The conventional or classic drug discovery approach has been expensive, time-wasting, and complex (Wang et al. 2015). The growth in the field of computation, as well as the availability of varieties of computational chemistry software, has facilitated drug development and discovery process such as a structure-based approach to be less expensive, timely, and efficient (Macalino et al. 2020; Anusuya et al. 2016). Among the structure-based approaches, molecular docking is becoming interestingly reliable owing to its successful application in the identification of lead compounds as well as the screening of large compounds databases for potentially active compounds (Benmansour et al. 2016; Ul et al. 2016).
One of the most important strategies is structure-based drug design (SBDD) that has been molecular docking studies (Benmansour et al. 2016; Ul et al. 2016). Generally, the docking technique entails predicting ligand conformation (poses) and calculating the free binding energy of all poses using a score derived from knowledge-based potential. Docking analyses are particularly useful for predicting the protease's binding location as well as determining the binding affinities of drugs on protease structures (Meng et al. 2011).
The role of computer-aided drug discovery approaches ranged from virtual screening to estimation of drug-likeness (D-L), bioavailability, medicinal chemistry ADMET (Absorption/Distribution/Metabolism, Excretion/Toxicity) properties of the significant number of chemical compounds with high potential of being active and fulfilling the prerequisites of auspicious drugs (Daina et al. 2017).
Quantitative structure–activity relationship studies (QSARs), molecular docking, and pharmacokinetics and toxicity studies are only a few of the in silico methodologies employed in drug research and development. Correlations between chemical structures and their descriptors are statistically investigated in QSAR studies in order to detect correlations that could be used to predict biological activity (Vilar et al. 2008; Adawara et al. 2021). Molecular pharmacokinetics and toxicity evaluations give adequate information on features that influence a molecule's pharmacokinetics (Butina et al. 2002; Daina et al. 2017), and molecular docking simulation elucidates interactions between binding molecules (Macalino et al. 2020).
We aimed to explore this proficient, less expensive, and accurate approach of drug discovery to design an inhibitor of the DENV NS5 protease with high potency than the lead compound identified in our previous work (Adawara et al. 2021) through the structure-based design method through the modification of the lead as well as predicting their D-L, bioavailability, ADMET, and medicinal chemistry to avoid failure after development or advance discovery state and adverse effect. We hope that the outcome of this work could explain the basis for the better activity of DENV inhibitor of such class of compounds as well as provide information that could ease the design and synthesis of effective, less toxic, and good pharmacokinetic DENV inhibitors that could aid it the treatment of infections caused by Flavivirus.
Methods
Data set
The data used in this study is the lead compounds identified from our previous work (Adawara et al. 2021) presented in Fig. 1 in which a quantitative structure–activity study was carried out as well as virtual screening for potential lead identification. In furtherance for the study, in which compound 34 ((E)-5-(2-(5-bromothiophen-2-yl) vinyl)-3-(3-chlorophenyl)-1, 2, 4-oxadiazole) was hinted as the lead, hence this study was initiated. Compound 34 has been reported to possess a good docking score of − 19.10 kcal/mol using ICM pro, likewise favourably interacted with the active site amino acid residues of DENV-2 NS-5 protease (Adawara et al. 2021).

Compound 34 was also found within the applicability domain of the model developed which entails its similarity with other derivatives as well as the predictivity of its inhibitory activity by the model. The lead compound has been reported elsewhere to have better pharmacological activity in a cell culture assay (Benmansour et al. 2016).
Insilco design of the hypothetical compounds
Using the information obtained from the lead with its interaction with the biochemical target (PDB: 5K5M), the analogues of the lead compound were designed through replacement, addition, or the removal of side-chain atoms in the structure of the selected lead compound (Benmansour et al. 2016; Adawara et al. 2021). The substitution was carried out at phenyl pharmacophore of the lead with methylamine hydroxyl and methoxy groups.
After this, a methodical substitution on the phenyl pharmacophore of the lead resulted in the lead analogues with modifications at the terminal of the chlorophenyl pharmacophore. Towards this end, ten analogues of the lead compound were designed. The 2-dimensional chemical structures (2D) of the compounds were drawn by the use of Chemdraw (Li et al. 2004). The ten drawn designed compounds are presented in Table 1.
Pre-docking preparation of designed compounds and target protease
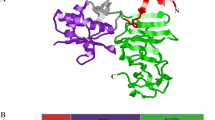
After drawing the chemical structure of the hypothetical compounds, we further subject them to energy minimization to obtain them in their best conformation and converted them to PDB readable file format. The energy minimization was achieved using the B3LYP (Lee–Yang–parr hybrid functional) level using 6–31G* as the basis set of the Density Function Theory technique implemented in Spartan 14 (Hehre and Huang 1995). The DENV protease earlier used as the target in our previous work was used in this as the target which was obtained from http://www.rcsb.org/pdb protein data bank (PDB ID: 5K5M). The protease was acquired in complex with other compounds (potential target inhibitor) (Lim et al. 2016). The preparation of the protease involved the deletion of heteroatoms and water molecules and the addition of hydrogen which has been described (Adawara et al. 2021). The prepared 3D structure of the protease is presented in Fig. 2.

Prepared 3D structure of the therapeutic target (PDB: 5K5M) 2.01 Å
Docking calculation and virtual screening
We accomplished molecular docking calculation for the estimation of the binding mode/affinity between the DENV Serotype 2 RNA Dependent RNA Polymerase NS-5 receptor (PDB ID: 5K5M) presented in Fig. 2 and the designed compound (ligand) presented in Table 1 with the aid of the Molsoft IC-M-Pro (Neves et al. 2012) to obtain the binding mode of the designed compounds to the NS-5 protease.
Protease preparation before the docking calculations, as well as the binding interaction mode visualization, was done using Discovery Studio 2017 (DST) (Biovia 2017).
Due to the absence of any specifically approved drug for the treatment of dengue virus disease, in this study, we well-thought-out Fenretinide (4-HPR (N-(4-hydroxyphenyl)-retinamide) based on our previous study where it was considered as the standard inhibitor (Adawara et al. 2021) and S-adenosyl-l-homocysteine (SAH) as standard inhibitors since Fenretinide was reported elsewhere (Behnam et al. 2016) to be involved in inhibiting the DENV NS5 polymerase as well as demonstrated activity in the prevention of viral replication against all serotypes in cell culture as well as mouse model, whereas, S-adenosyl-l-homocysteine (SAH) has been earlier considered as Flavivirus NS5 inhibitor (Behnam et al. 2016). Alongside the Ferentinide and SAH, Ribavirin was also taken into account as standard.
Additionally, to substantiate our evidence, the co-crystal ligand obtained in complex with the protease from the PDB was removed, optimized, and re-docked with the protease. Its interaction with the protease before preparation and after the docking was viewed using the DST.
The docking scoring function is based on force-field interaction energy terms and is a function of the free binding free affinity between a ligand and a protease. The lower the score, the better the ligand's chances of becoming a good binder (Adawara et al. 2020).
In silico ADMET predictions of the designed DENV inhibitors
The design compounds after being successfully docked were as well subjected to ADMET and D-L evaluation. The ADMET and D-L predictions of the designed compounds were accomplished using the Swiss-ADME and pkCSM web tools (Daina et al. 2017; Pires et al. 2015).
Results
The molecular docking results for the designed compounds
The results of the molecular docking calculations binding affinity scores (kcal/mol) of the designed compounds (Table 1) with protease using the Molsoft IC-M-Pro software are presented in Table 2. The binding score designates how strongly bound the interaction of the compounds with the biochemical target (PDB ID: 5K5M) is Yokokawa et al. (2016). It is expressed in the unit of kcal/mol and the lower the numeric value the better the interaction. The molecular docking interactions of the designed compounds and some selected standard inhibitors with the individual amino acid involving various interactions type and the nature of such interactions as well as their bond distances are illustrated in Table 3. The visualized 2D docking interactions of the ligand-protease complexes for the designed compounds and standard inhibitors using DST are shown in Additional file 1: Figs. (SF) 1a–j and 3a–f, whereas those of the co-crystal ligand of the protease before and after docking are presented in Additional file 1: Fig. SF2a, b, respectively.
Designed compounds’ predicted ADMET
The results of the predicted D-L, pharmacokinetics, and ADMET parameters of the designed compounds (Table 1) are presented in Tables 4 and 5. Table 4 illustrates the parameters suggested by Lipinski for D-L and oral bioavailability, gastrointestinal adsorption, PAINS alert synthetic accessibility, and bioavailability score. Table 5 presents some selected toxicity profiles of the designed compounds.
Discussion
Molecular docking of the designed compounds with the target
The designed compounds after optimization were subjected to molecular docking simulation to validate the improvement in the interaction of the lead compound for a better inhibition of the DENV NS-5 protease. The results of the docking scores of the designed compounds (34a, 34b, 34c, 34d, 34e, 34f, 34g, 34h, 34i, and 34j) obtained from the Molsoft IC-M-Pro are presented in Tables 2 and 3 shown to be − 26.54, − 29.612, − 22.652, − 23.644, − 23.594, − 19.992, − 23.943, − 19.292, − 20.121, − 19.091, − 22.347 kcal/mol, respectively.
The interaction of the designed compounds with protease amino acid residues indicating the individual residues’ interaction distance as well as the nature and the type of the interactions is presented in Table 3. From Table 2, it could be observed that the docking score of the designed compounds ranged between − 19.091 and − 29. 61 kcal/mol; the lead compound has been reported to have a binding/docking score of − 19.10 kcal/mol (Adawara et al. 2021). The designed compound-protease interactions, as well as those of the standard inhibitors, are illustrated in Fig. 3a–d and Additional file 1: SF1.

2D docking interaction of some selected design compounds (34a and 34b) (a, b) and standard inhibitors (SAH and Ribavirin) (c, d) with the DENV NS-5 protease (PDB ID: 5K5M)
Compound 34a (binding score − 26.54 kcal/mol) was observed to form three conventional hydrogen bonds (C HB) with SER763, CYS780, and SER885 (1.852, 1.799, and 2.084 Å) amino acid residues and one carbon-hydrogen bond (C–H-B) with ARG773 amino acid residue. About eight hydrophobic interactions were also formed with TYR882, ASN777, TRP833, TRP833, TYR882, and HIE786 A: CYS780 and MET809 residues through Pi–Sulfur, Pi–Lone Pair, Pi–Pi Stacked, Pi–Pi Stacked, Pi–Pi T-shaped, Pi–Alkyl, Pi–Alkyl, and Pi–Alkyl, respectively. The conventional hydrogen bond formation in compound 34a involved oxygen and nitrogen at position 2 of the oxadiazole core, as well as the –HO group at the para position in which it interacted with SER885 amino residue where it acts as hydrogen bond donor.
Compound 34b with the best docking score of − 29. 61 kcal/mol was observed to participate in the interactions involving four C HB with ASN777, TRP833, SER885, SER885 (1.858, 2.164, 1.836, and 2.123 Å) amino acid residues where the two –OH group at the ortho and meta position of the phenyl ring both donated hydrogen to SER885 residue, while the oxygen of the –OH group at the para position of the phenyl ring and the nitrogen at position 2 of the oxadiazole account for the other two C HB interactions involving ASN777 and TRP833 residues where they act as donors. There was no C–H-B interaction observed in compound 34b, other than the four C BH and nine hydrophobic interactions involving MET809, MET809, CYS780, TYR882, ASN777, TRP833, TRP833, TYR882, and HIE786 amino acid residues through Pi–Donor Hydrogen Bond, Pi–Sigma, Pi–Sulfur, Pi–Sulfur, Pi–Lone Pair, Pi-Pi Stacked, Pi–Pi Stacked, Pi–Pi Stacked, and Pi-Alkyl.
Ribavirin formed eight favourable conventional hydrogen bond interactions involving LYS756, SER763, CYS780, CYS780, ASN777, GLN760, GLN760, and THR806 (1.690, 1.853, 2.536, 2.467, 2.799, 2.896, 2.208, and 2.554 Å) were observed as well as five C–H-B with THR806, SER785, THR806, GLU807, GLN760 amino acid residue, whereas the MET809, CYS780, MET809 residues were involved in hydrophobic interactions through Pi–Donor Hydrogen Bond, Pi–Alkyl, and Pi–Alkyl.
SAH has the highest hydrogen bond interaction energy of − 10.4372 and formed six C HB interactions with LYS756, ASN777, CYS780, MET809, GLN760, GLN760 (2.325, 1.871, 1.840, 2.925, 2.099, 2.047 Å) amino acid residues, and formed seven C–H-B interactions with SER763, ASP808, GLN760, SER785, THR806, ASP808, HIE786, with other two hydrophobic interaction TYR882, HIE786 residues through Pi–Sulfur, Pi–Pi T-shaped, respectively. The higher hydrogen bond energy observed in SAH could be due to majorly C–H-B interaction because it has the highest amount of C–H-B interaction but a lower binding score of − 16.536 kcal/mol.
The co-crystal ligand of the protease demonstrated a binding score of − 25.0433 kcal/mol (Table 2), but despite having such a higher binding score was observed to form some unfavourable bond (Additional file 1: Fig. SF2b) which entail instability of the complex. The 2D binding interactions of the native ligand-protease presented in Additional file 1: Fig. SF2 were viewed before and after docking (Additional file 1: Fig. SF2a, b) to understand the significance of optimizing the co-crystal ligand in terms of how it interacts with the protease.
In summary, the hydroxyl group of the phenyl moiety, as well as those of the methylamine and methoxy groups, formed conventional hydrogen bond interaction with some important amino acid residues of the protease. This observation could be responsible for the higher hydrogen bond energy interactions obtained for the designed compounds which are important for the ligand-protease complex stability. More so, the nitrogen at position two of the oxadiazole core was observed to be stabilized through conventional hydrogen in all the designed compounds, this highlights the importance of nitrogen at position two in conferring the stability of the ligand protease complexes.
The compounds all formed favourable interactions with the protease which entails the good potential of the compounds as inhibitors. Among the designed compounds, compounds 34a and 34b showed remarkable docking scores far much better than the lead as well as the standard inhibitor, although, none of the inhibitors had a docking score close to the lead talk more of the improved derivatives of the lead.
The stabilization of the complexes of the designed compound-protease was majorly through conventional hydrogen bond and hydrophobic bond interactions involving residues at the allosteric sites of the protease. All the designed compounds have a better hydrogen bond energy (− 3.402 to − 9.0128 kcal/mol) than the template (− 3.1 kcal/mol) which entails favourable interactions with the protease than the template and were all found to be in a similar manner as the standard inhibitors. This could bring about better stability of the complexes, hence better inhibitory activity.
D-L, pharmacokinetics, and ADME prediction of the designed compounds
D-L of any potential drug candidate is essential in the drug development process. The D-L properties for potential drug candidate proposed by Lipinski implemented in the Swiss-ADME web tool were utilized. The obtained D-L parameters are presented in Table 4. It could be observed that all the designed compounds perfectly obeyed the rules suggested by Lipinski characterized by their molecular weight of not less than 500, logP value of not greater than 5, hydrogen-bond donors of not greater than 5; hydrogen-bond acceptors of not greater than 10, and topological polar surface area (TPSA) of less than 140 recommended by Lipinski (Lipinski 2016; Daina et al. 2017), from the predicted properties presented in Table 4, it could also be seen that our designed compounds passed all Lipinski’s rule of five which also suggests good D-L and oral bioavailability (Lipinski 2016).
The estimation of the ease of synthesis (synthetic-accessibility) of bioactive compounds possessing drug-likes-ness is an essential need in the drug discovery process (Ertl and Schuffenhauer 2009).
Other valuable information obtained from the ADME evaluation presented in Table 4 includes the gastro-intestinal adsorption (GIA), pan-assay interference compounds (PAINS) alert, bioavailability score, and synthetic accessibility. The designed compounds could all be seen to possess high GIA except the standards inhibitors considered, which entail easy and favourable GIA by the designed compound.
The designed compounds were predicted to possess no PAINS alert except compound 34b with one PAINS alert which depicts the true activity of the compounds in the biochemical assay (Baell and Holloway 2010).
The bioavailability scores of the designed compounds, as well as those of the standard all, fall within the range of active category as compounds with bioavailability scores in this range, are classified as highly active (Ertl and Schuffenhauer 2009; Mishra et al. 2016). The compounds have all demonstrated the ease of synthesis evidenced by their synthetic accessibility score of 3.27–3.83 which are lower than those of the standards since the smaller the value, the easier a chemical compound could be synthesized (Ertl and Schuffenhauer 2009).
The drug-metabolizing capacity of CYP450 enzymes, clinically relevant CYP450 genetic polymorphisms, cytochrome P450 CYP-1A2, CYP-2C9, CYP-2C19, and CYP-2D6 were also evaluated. The compounds including the standard are all non-Pgp substrate, as well as CYP2D6, a CYP3A4 inhibitor, whereas only compound 34j was found to be non-inhibitor of CYP-2C9, respectively (Table 4) (Hollenberg 2002; Serretti et al. 2009).
The toxicity of the designed compounds was assessed using the pkCSM webpage tool, with the results presented in Table 5. The results revealed that the designed compounds all had no AMES toxicity, no skin sensitization, and were all non-inhibitors of the human ether-a-go-go-related gene (hERG) cardiovascular toxicity, making them safer. Except for compounds 34c, all of the compounds' hepatotoxicity potential was assessed to be positive. The proposed compounds' Oral Rat Acute Toxicity (LD50) ranged from 1.894 to 2.674, while SAH Ribavirin and Fenretinide had 2.485, 1.481, and 2.696, respectively, indicating that they are in the same range as Ribavirin and are even safer. Based on the toxicity profile of the developed compounds, it may be reasonable to classify them as non-toxic, and they have been demonstrated to have good D-L.
Conclusions
Through structural modification of the lead (compound 34) from our previous study, and subsequent molecular docking, D-L, ADME, and toxicity evaluation of the hypothetically active compounds, we were able to design highly potent and less toxic dengue virus serotype 2 inhibitors targeting the NS-5 protease using a structure-based drug design approach from a template of the class of 1, 2, 4-oxadiazole derivative identified in our previous work. Ten derivatives of the lead drug were developed and found to bind better than the lead compound, with a molecular docking score of − 19.19 to − 29.3 kcal/mol, outperforming the template, the standard inhibitor. All the compounds had better hydrogen bond interaction than the template which entails better interaction with the biological target. The designed compounds' D-L, ADME, and toxicity estimations demonstrate good D-L and desirable ADME parameters, but no AMES toxicity was detected among all the designed compounds, and other toxicity aspects analyzed suggested a relatively safe drug. The proposed compounds have a good possibility of being made into an anti-DENV medication. This research also provides a foundation for further synthesis of such potent derivatives to create novel treatment options for treating dengue virus infections at a low cost and on time.
Availability of data and materials
Not applicable.
Abbreviations
- DENV:
-
Dengue virus
- NS:
-
Non-structural protein
- PDB:
-
Protein data bank
- ADMET:
-
Adsorption/distribution/metabolism/excretion/toxicity
- D-L:
-
Drug-likeness
- SBDD:
-
Structure-based drug design
- DST:
-
Discovery studio
- SAH:
-
S-Adenosyl-l-homocysteine
- C HB:
-
Conventional hydrogen bond
- C–H-B:
-
Carbon hydrogen bond
- HB:
-
Hydrogen bond
- GIA:
-
Gastro-intestinal adsorption
- PAINS:
-
Pan-assay interference compounds
- TPSA:
-
Topological polar surface area
- 2D:
-
2-Dimensional
- SF:
-
Supplementary figure
References
Adawara SN, Mamza P, Gideon SA, Ibrahim A (2020) Anti-dengue potential, molecular docking study of some chemical constituents in the leaves of isatis tinctoria. Chem Rev Lett 3(3):104–109
Adawara SN, Shallangwa GA, Mamza PA, Ibrahim A (2021) In silico studies of oxadiazole derivatives as potent dengue virus inhibitors. Chem Afr 4:1–8
Anusuya S, Gromiha MM (2019) Structural basis of flavonoids as dengue polymerase inhibitors: insights from QSAR and docking studies. J Biomol Struct Dyn 37(1):104–115
Anusuya S, Velmurugan D, Gromiha MM (2016) Identification of dengue viral RNA-dependent RNA polymerase inhibitor using computational fragment-based approaches and molecular dynamics study. J Biomol Struct Dyn 34(7):1512–1532
Baell JB, Holloway GA (2010) New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem 53(7):2719–2740
Batool F, Saeed M, Saleem HN, Kirschner L, Bodem J (2021) Facile synthesis and in vitro activity of N-substituted 1, 2-benzisothiazol-3 (2H)-ones against dengue virus NS2BNS3 protease. Pathogens 10(4):464
Behnam MA, Nitsche C, Boldescu V, Klein CD (2016) The medicinal chemistry of dengue virus. J Med Chem 59(12):5622–5649
Benmansour F, Eydoux C, Querat G, de Lamballerie X, Canard B, Alvarez K, Guillemot JC, Barral K (2016) Novel 2-phenyl-5-[(E)-2-(thiophen-2-yl) ethenyl]-1, 3, 4-oxadiazole and 3-phenyl-5-[(E)-2-(thiophen-2-yl) ethenyl]-1, 2, 4-oxadiazole derivatives as dengue virus inhibitors targeting NS5 polymerase. Eur J Med Chem 109:146–156
Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O, Myers MF (2013) The global distribution and burden of dengue. Nature 496(7446):504–507
Biovia DS, DSME R. San Diego: Dassault Systèmes (2017)
Butina D, Segall MD, Frankcombe K (2002) Predicting ADME properties in silico: methods and models. Drug Discov Today 7(11):S83–S88
Daina A, Michielin O, Zoete V (2017) SwissADME: a free web tool to evaluate pharmacokinetics, D-L and medicinal chemistry friendliness of small molecules. Sci Rep 7(1):1–3
Ertl P, Schuffenhauer A (2009) Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J Cheminform 1(1):1–1
Hehre WJ, Huang WW (1995) Chemistry with computation: an introduction to SPARTAN. Wavefunction. Inc, Irvine.
Hollenberg PF (2002) Characteristics and common properties of inhibitors, inducers, and activators of CYP enzymes. Drug Metab Rev 34(1–2):17–35
Li Z, Wan H, Shi Y, Ouyang P (2004) Personal experience with four kinds of chemical structure drawing software: review on ChemDraw, ChemWindow, ISIS/Draw, and ChemSketch. J Chem Inf Comput Sci 44(5):1886–1890
Lim SP, Noble CG, Seh CC, Soh TS, El Sahili A, Chan GK, Lescar J, Arora R, Benson T, Nilar S, Manjunatha U (2016) Potent allosteric dengue virus NS5 polymerase inhibitors: mechanism of action and resistance profiling. PLoS pathogens 12(8):e1005737
Lipinski CA (2016) Rule of five in 2015 and beyond: Target and ligand structural limitations, ligand chemistry structure and drug discovery project decisions. Adv Drug Deliv Rev 101:34–41
Macalino SJ, Billones JB, Organo VG, Carrillo MC (2020) In silico strategies in tuberculosis drug discovery. Molecules 25(3):665
Megawati D, Masyeni S, Yohan B, Lestarini A, Hayati RF, Meutiawati F, Suryana K, Widarsa T, Budiyasa DG, Budiyasa N, Myint KS (2017) Dengue in Bali: clinical characteristics and genetic diversity of circulating dengue viruses. PLoS Negl Trop Dis 11(5):e0005483
Meng XY, Zhang HX, Mezei M, Cui M (2011) Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des 7(2):146–157
Mishra SS, Sharma CS, Singh HP, Pandiya H, Kumar N (2016) In silico ADME, Bioactivity and toxicity parameters calculation of some selected anti-tubercular drugs. Int J Pharmacol Phytopharmacol Res 6:77–79
Neves MA, Totrov M, Abagyan R (2012) Docking and scoring with ICM: the benchmarking results and strategies for improvement. J Comput Aided Mol Des 26(6):675–686
Pires DE, Blundell TL, Ascher DB (2015) pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem 58(9):4066–4072
Serretti A, Calati R, Massat I, Linotte S, Kasper S, Lecrubier Y, Sens-Espel R, Bollen J, Zohar J, Berlo J, Lienard P (2009) Cytochrome P450 CYP1A2, CYP2C9, CYP2C19 and CYP2D6 genes are not associated with response and remission in a sample of depressive patients. Int Clin Psychopharmacol 24(5):250–256
ul Qamar MT, Kiran S, Ashfaq UA, Javed MR, Anwar F, Ali MA, Gilani AuH (2016) Discovery of novel dengue NS2B/NS3 protease inhibitors using pharmacophore modeling and molecular docking-based virtual screening of the zinc database. Int J Pharmacol 12(6):621–632
Vilar S, Cozza G, Moro S (2008) Medicinal chemistry and the molecular operating environment (MOE): application of QSAR and molecular docking to drug discovery. Curr Top Med Chem 8(18):1555–1572
Wang T, Wu MB, Lin JP, Yang LR (2015) Quantitative structure-activity relationship: promising advances in drug discovery platforms. Expert Opin Drug Discov 10(12):1283–1300
Wilder-Smith A, Gubler DJ, Weaver SC, Monath TP, Heymann DL, Scott TW (2017) Epidemic arboviral diseases: priorities for research and public health. Lancet Infect Dis 17(3):e101–e106
Yokokawa F, Nilar S, Noble CG, Lim SP, Rao R, Tania S, Wang G, Lee G, Hunziker J, Karuna R, Manjunatha U (2016) Discovery of potent non-nucleoside inhibitors of dengue viral RNA-dependent RNA polymerase from a fragment hit using structure-based drug design. J Med Chem 59(8):3935–3952
Acknowledgements
The authors gratefully acknowledged the technical effort of Dr. Tukur Mohammed of the Department of Chemistry, Ahmadu Bello University, Zaria.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
SNA designed and wrote the manuscript, GAS, PAM, and IA supervised and carried out the statistical analysis. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
: Figure SF1. 2D binding interactions between the receptor and all the designed compounds (34a–34j). Figure SF2. 2D binding interactions of the co-crystal ligand of the protease view before (a) and after (b) molecular docking with the receptor, respectively
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Adawara, S.N., Shallangwa, G.A., Mamza, P.A. et al. Chemoinformatic design and profiling of some derivatives of 1, 2, 4-oxadiazole as potential dengue virus NS-5 inhibitors. Bull Natl Res Cent 46, 65 (2022). https://doi.org/10.1186/s42269-022-00755-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42269-022-00755-7