
Abstract
Background
Due to versatility in reaction catalyzed by peroxidases, they have potential applications in different areas in the health sciences, food industry, and diagnostic purposes. Therefore, the aim of this study is to investigate the properties of peroxidase from ginger to be meeting the perquisites of several applications.
Results
The cationic peroxidase (GPII) was purified to homogeneity by anion exchange chromatography using DEAE–Sepharose column followed by cation exchange chromatography using CM–Sepharose column and finally Sephacryl S-200 column. The molecular mass of GPII was 42 kDa. GPII shows oxidizing activity with several phenolic compounds by using H2O2 as the second substrate. The natural plant phenolic compounds as pyrogallol, catechol, and guaiacol were found to be excellent electron donors for the enzyme compared to other phenolic compounds. GPII exhibited Km values of 3.1 and 7.1 mM and Vmax values of 0.6 and 0.31 units/assay using H2O2 and guaiacol as substrates, respectively. The enzyme exhibited maximal peroxidase activity at broad pH’s 6.0–7.5 and 50 °C. GPII was thermal stable up to 50 °C and retained 66% of its activity at 70 °C after 1 h incubation. The GPII activated by most divalent cations tested and inhibited by Hg2+ and Cu2+ cations.
Conclusion
PGII could be used in several applications due to its catalytic properties, thermal stability, broad pH, and acting on several phenolic compounds.
Similar content being viewed by others
Background
Peroxidases (EC.1.11.1.x) are decomposing hydrogen peroxide (H2O2) to oxidize a wide variety of phenolic compounds as well as non-phenolic compounds (Pandey et al. 2017). The non-animal plant peroxidases, belonging to Class III peroxidase, are enzymes that participate in diverse physiological and biochemical functions in higher plant cells and are, therefore, interesting objects of current biochemical research (Has-Schön et al. 2005; Pandey et al. 2017). They are implicated in various vital processes in vivo include cell wall edification, cellular growth, differentiation and development, in hormone catabolism, lignin polymerization, suberization, fruit growth and ripening, ethylene biosynthesis, plasma membrane redox systems and the generation of H2O2, auxin metabolism, senescence, and defense mechanisms against abiotic and biotic stress (Chen et al. 2012; Mohamed et al. 2014; Aslmoshtaghi and Shahsavar 2016; Galende et al. 2016; Julião et al. 2016).
Due to versatility in reaction catalyzed by peroxidases, and their ubiquitous nature, they have potential applications in different areas in the health sciences, food industry, and diagnostic purposes (Pandey et al. 2017; Abdel-Aty et al. 2018). Peroxidases are implicated in various applications in detoxification of recalcitrant organopollutants, decolourization of textile effluents, in bioremediation, polymer synthesis, paper and pulp industry, in development of biosensor, diagnosis kits, etc. (Lavery et al. 2010; Bhatti et al. 2012; Kalsoom et al. 2013; Sarvamangala 2014; Nouren et al. 2015; Tabassum et al. 2015; Mohamed et al. 2017; Moubasher et al. 2017).
Plants as sources of enzymes have become a promising field of study, because of the simplicity and ease of obtaining this biological material (Julião et al. 2016). Despite the variety of plant peroxidase sources, there is no previous study on peroxidase from Zingiber officinale commonly known as ginger, originated in the Indo-Malayan region, is now widely distributed across the tropics of Asia, Africa, America and Australia (Yeh et al. 2014). Ginger belonging to the family Zingiberaceae, is widely consumed as a spice used for over 2000 years in various foods and beverages (Chari et al. 2013). Ginger is reported to have several beneficial pharmacological effects (hypoglycemic, insulinotropic, and hypolipidemic) on health in humans (Huang et al. 2004) and in experimental animals (Akhani et al. 2004; Kondeti et al. 2011). Therefore, the aim of this study is to investigate the properties of peroxidase from ginger to be meeting the perquisites of several applications.
Methods
Plant
Fresh ginger (Zingiber officinale) was obtained from the local market, Cairo, Egypt.
Peroxidase assay
Peroxidase activity was determined by the method of Miranda et al. (1995). One milliliter of reaction mixture including 40 mM guaiacol, 8 mM H2O2, 20 mM sodium acetate buffer, pH 5.5, and 100 μl of enzyme. The absorbance was measured at 470 nm. One unit of peroxidase activity is defined as the amount of enzyme that increases the optical density of 1.0 per min under standard assay conditions.
Protein determination
Protein concentration was quantified by the method of Bradford (1976) using bovine serum albumin as standard.
Crude enzyme extract
Two grams of fresh ginger was homogenized with 20 mM Tris-HCl buffer, pH 7.2 using mortar. The homogenate was centrifuged at 12,000 rpm for 10 min in cooling centrifuge. The supernatant was designated as a crude enzyme extract and stored at − 20 °C until further analysis.
Purification of peroxidases from ginger
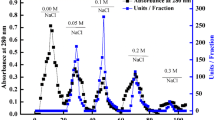
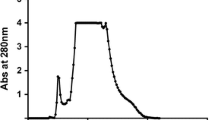
Unless otherwise stated, all purification steps of peroxidases from ginger were carried out at 4 °C. The crude enzyme extract was fractionated on DEAE-Sepharose column (10 × 1.2 cm i.d.) pre-equilibrated with 20 mM Tris-HCl buffer, pH 7.2 and eluted with a stepwise gradient of NaCl (0.0–0.4 M) in the same buffer. Fractions of 5.0 ml were collected at 4 °C and a flow rate of 30 ml/h. The most of peroxidase activity was eluted at 0.0 M NaCl and the remainder of peroxidase activity was eluted by different concentrations of NaCl. The peroxidase eluted at 0.0 M NaCl was dialyzed against 20 mM sodium acetate buffer, pH 5.5 and applied on CM-Sepharose column (10 × 1.2 cm i.d.) pre-equilibrated with the same buffer. The adsorbed proteins were eluted with a stepwise gradient of NaCl (0.0 to 0.4 M) in the same buffer at a flow rate of 30 ml/h, and 5.0 ml fractions were collected. The most peroxidase activity was eluted at 0.1 M NaCl and designated as GPII. The GPII was applied on Sephacryl S-200 and the enzyme eluted by 20 mM sodium acetate buffer, pH 5.5 at a flow rate of 20 ml/h, and 3.0 ml fractions were collected.
Molecular weight measurement
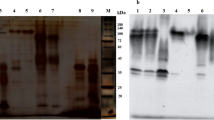
Molecular weight was determined by gel filtration technique using Sephacryl S-200 column. The column (93 × 1.6 cm i.d.) was calibrated with cytochrome C (12,400), carbonic anhydrase (29,000), bovine serum albumin (67,000), alcohol dehydrogenase (150,000), and β-amylase (200,000). Dextran blue (2,000,000) was used to determine the void volume (Vo). Subunit molecular weight was estimated by SDS-polyacrylamide gel electrophoresis (Laemmli 1970). SDS-denatured phosphorylase b (94,000), bovine serum albumin (67,000), ovalbumin (43,000), carbonic anhydrase (30,000), soybean trypsin inhibitor (20,000), and α-lactalbumin (14,200) were used for calibration.
Characterization of GPII
The Km values of GPII were determined from Lineweaver–Burk plots by using different concentrations of H2O2 and guaiacol. The optimum pH of GPII was examined using 20 mM sodium citrate buffer (pH 4.0–4.5), sodium acetate buffer (pH 4.5–6.0), sodium phosphate buffer (pH 6.5–8.0), and Tris–HCl buffer (pH 8.0–9.0), respectively. The optimum temperature of GPII was determined by incubating the reaction mixture at different temperatures ranging from 25 to 80 °C. The effect of temperature on the enzyme stability was examined by pre-incubating the enzyme for 30 min in different temperatures ranging from 25 to 80 °C prior to substrate addition, followed by cooling in an ice bath, and the remaining activity was measured. The effect of metal ions on the peroxidase activity was also determined. The enzyme was pre-incubated for 30 min at 37 °C with some metal ions individually at the final concentration indicated prior to substrate addition and the remaining activity was measured.
Statistical analysis
The data were statistically analyzed by a one-way ANOVA. The data were considered means ± S.E. (n = 3).
Results
The purification of peroxidase from ginger was summarized in Table 1. The crude enzyme extract was fractionated on DEAE-Sepharose column (10 × 1.2 cm i.d.). The most of peroxidase activity was eluted at 0.0 NaCl and five peaks with low-level peroxidase activity were eluted by different concentrations of NaCl (Fig. 1). The peroxidase eluted at 0.0 NaCl was applied to CM-Sepharose column (10 × 1.2 cm i.d.). One peak of protein with low peroxidase activity was eluted at 0.0 M NaCl and designated as PGI. The most peroxidase activity was eluted at 0.1 M NaCl and designated as cationic GPII (Fig. 2). The GPII with high peroxidase activity was applied on Sephacryl S-200, where GPII retained 61.2% of its initial activity with specific activity 13,136 units/mg protein and 11.8 purification fold (Fig. 3). In the assessment of homogeneity using SDS-PAGE, the electrophoretic profile of proteins showed that the purified GPII enzyme was migrated as a single band of molecular weight of 42 kDa (Fig. 4).

A typical elution profile for the chromatography of ginger peroxidase on DEAE-Sepharose column (6 × 1.6 cm i.d.) previously equilibrated with 20 mM Tris-HCl buffer, pH 7.2, at a flow rate of 30 ml/h and 5 ml fractions

A typical elution profile for the chromatography of 0.0 M fraction of DEAE-Sepharose of ginger peroxidase on CM-sepharose column (6 × 1.6 cm i.d.) previously equilibrated with 20 mM sodium acetate buffer, pH 5.5, at a flow rate of 30 ml/h and 5 ml fractions

A typical elution profile for the chromatography of GPII CM-Sepharose fraction on a Sephacryl S-200 column (90 × 1.6 cm i.d.) previously equilibrated with 50 mM sodium acetate buffer, pH 5.5, at a flow rate of 20 ml/h and 3 ml fractions

SDS-PAGE for molecular weight determination for Sephacryl S-200 GPII. 1. Standard proteins, 2. Sephacryl S-200 GPII
The activity of the purified GPII towards various substrates was summarized in Table 2. The activity towards guaiacol, considered standard substrate for class III peroxidase, was taken as 100%. Substrate assay profile of the GPII enzyme revealed that the enzyme was highly active towards pyrogallol and catechol with relative activities % of 140 and 118, respectively. However, substrates as o-dianisidine, 4-aminoantipyrin, o-phenylenediamine, and m-phenylenediamine are oxidized by GPII at slower rates with relative activities % of 32, 34, 25 and 20, respectively. The Lineweaver-Burk plots relating ginger GPII reaction velocities to H2O2 and guaiacol were used for measuring the Michaelis-Menton kinetics (Km and Vmax) (Fig. 5). The Km and Vmax obtained for the purified GPII were 3.1 mM and 0.6 units/assay and 7.1 mM and 0.31 units/assay for H2O2 and guaiacol, respectively.

Lineweaver-Burk plots relating ginger GPII reaction velocities to H2O2 and guaiacol
The effect of pH on the purified GPII was investigated at different pH’s ranged from 4.0 to 9.0. The optimum pH for GPII was found at broad pH’s 6.0–7.5 (Fig. 6). The optimum temperature for the purified GPII was 50 °C (Fig. 7). The enzyme retained 65 and 40% of its residual activity at 70 and 80 °C, respectively. GPII was thermal stable up to 50 °C and retained 75 and 55% of its activity at 70 and 80 °C for 1 h incubation, respectively (Fig. 8).

pH optimum of ginger GPII. The reaction mixture contained in 1.0 ml: 8 mM H2O2, 40 mM guaiacol, a suitable amount of enzyme, and 20 mM sodium citrate buffer (pH 4.0–4.5), sodium acetate buffer (pH 4.5–6.0), sodium phosphate buffer (pH 6.5–8.0), and Tris–HCl buffer (pH 8.0–9.0). The points are presented as mean ± S.E. (n = 3)

Temperature optimum of ginger GPII. The enzyme activity was measured at various temperatures under standard assay conditions. The points are presented as mean ± S.E. (n = 3)

Effect of temperature on the thermal stability of ginger GPII. The enzyme was preincubated at various temperatures for 1 h prior to substrate addition, followed by cooling in an ice bath. Activity at zero time was taken as 100% activity. The points are presented as mean ± S.E. (n = 3)
The effect of various divalent metal cations on the activity of GPII is shown in Table 3. Most of the divalent cations as Ca2+, Ba2+, Zn2+, Mg2+, Fe2+, and Mn2+ activated the purified GPII, while the Hg2+ and Cu2+ cations were a strong inhibitor for GPII even at 2 mM, but this effect was more pronounced at 5 mM.
Discussion
The purification of peroxidase from ginger showed several isoenzymes of peroxidases. The cationic GPII binding on CM-Sepharose possessed the highest peroxidase activity. The specific activity of GPII (13,136 units/mg protein) was higher than that reported for commercial horseradish peroxidase (250 units/mg solid) as a product from Simgma Company. The presence of several peroxidases in ginger proved that these enzymes belonging to the class-III peroxidase gene family which had multiple isoenzymes like several plant species (Boucoiran et al. 2000; Tognolli et al. 2002; Mohamed et al. 2011b). In this gene family, peroxidases had several roles such as seed germination, lignin formation, cell elongation, and stress defense (Shigeto and Tsutsumi 2015; Abdel-Aty et al. 2019). The molecular weight of purified GPII (42 kDa) on SDS-PAGE is compatible with that obtained by Sephacryl S-200 column. The obtained molecular weight was in accordance with peroxidases from Panaeolus sphinctrinus (42 KDa), horseradish roots (40 kDa), and chick pea (39 kDa) (Heinzkill et al. 1998; Lavery et al. 2010; Bhatti et al. 2006).
Peroxidases catalyzed the oxidation of a wide assortment of electron donor substrates; such as phenols, aromatic amines, thioanisoles, and iodide (Mirazizi et al. 2016). The different relative activities observed for GPII may be accounted for the structural differences between different substrates. Regarding the chemical nature, GPII showed the highest oxidative capacity towards phenolic compounds containing two and three hydroxy groups in their chemical structures like catechol and pyrogallol manifests a moderate activity against phenolic compounds having one hydroxyl group as guaiacol. Similarly, peroxidases from Cucumis sativus had a high affinity towards pyrogallol and guaiacol (Battistuzzi et al. 2001). On the obverse side, the GPII was poorly active towards aromatic amines such as o-dianisidine, aminoantipyrin, and phenylenediamines. On the contrast, peroxidase POII from Adalia orange had low activity towards catechol compared with o-dianisidine and o-phenylenediamine (Mohamed et al. 2008). The results showed that the natural plant phenolic compounds as pyrogallol, catechol, and guaiacol were found to be excellent electron donors for GPII compared to other phenolic compounds. Therefore, GPII could be used as an antioxidant enzyme for preventing the oxidative damage by removal of the toxic H2O2, which oxidized the phenolic compounds.
While H2O2 theorize as a suicide substrate for peroxidases; the hydrogen donor substrate may keep the peroxidase active site safe from the inactivating action of H2O2 (Shukla et al. 2016). Therefore, the Km and Vmax of purified GPII was determined for H2O2 and guaiacol. A wide range of Km and Vmax values have been reported for other peroxidases from different plants. Taking guaiacol into consideration as a substrate, various Km and Vmax values were reported for Euphorbia tirucalli peroxidase (Km 4.13 mM and Vmax of 0.627 units/assay) (Shukla et al. 2016), umbu peroxidase (Km 6.83 mM and Vmax 4.16 units/assay) (Pinto et al. 2015) and peels of Citrus reticulata var. (Km 0.66 mM and Vmax of 380 units/assay) (Nouren et al. 2013). Using H2O2 as peroxidase substrate revealed Km/Vmax values of 1.8 mM/20 units/assay and 0.026 mM/0.8 units/assay for peroxidases extracted from Gongronema latifolium (Joy and Eze 2015) and garlic Allium sativum (Osuji et al. 2014), respectively.
GPII had optimum pH at broad pH’s 6.0–7.5. Similar pH optima were reported for peroxidases obtained from rice seedlings (Padiglia et al. 1995), strawberry fruits (Civello et al. 1995), and umbu plants roots (Pinto et al. 2015). A wide variability in the optimum temperature has been observed for peroxidases from various plants. The optimum temperature for the purified GPII was 50 °C. Different temperature optima were shown for peroxidases from rosemary leaves (40 °C) (Aghelan and Shariat 2015), white Spanish broom (30 °C) (Galende et al. 2016), and fingerroot (Boesenbergia rotunda (L.) Mansf.) (40 °C) (Shank et al. 2015). GPII was thermal stable up to 50 °C. Peroxidases from Cocos nucifera were thermal stable up to 50 °C for 1 h. (Balasubramanian and Boopathy 2013). Generally, peroxidase has been reported as the most thermostable enzymes in plants (Bhatti 2007; Khatun et al. 2012; Nouren et al. 2013).
The most of metal cations (Ca2+, Ba2+, Zn2+, Mg2+, Fe2+, and Mn2+) caused enhancement for the activity of GPII. Similarly, the Ca2+ has an imperative impact on molecular folding and usually demonstrates stimulatory effects on plant class III peroxidases (Ros-Barceló and Pomar 2002). Mathé et al. (2010) reported that Mn2+ as well as Ca2+ acts as prosthetic groups for particular peroxidases known for their specific catalytic action. The obtained results for Ca2+ and Fe2+ are in accordance with Galende et al. (2016). In contrast, Shukla et al. (2016) show a moderate to high % inhibition using Co2+ (29.6%), Ni2+ (36.6%), and Zn2+ (89.7%) on the activity of peroxidase from the latex of plant Euphorbia tirucalli. Fe2+ at a concentration of 2 and 5 mM activated the purified GPII with relative activity % of 155 and 215, respectively. Fe2+ plays an important role in the peroxidase-oxidase cycle that involves the formation of ferrous POD (Fe2+) and the inactive compound III (oxyperoxidase Fe6+) (Yamazaki and Yokota 1973). In contrast, Fe2+ highly inhibited the activity of partially purified peroxidase from fingerroot (Boesenbergia rotunda (L.) retaining 52% of the activity compared to the control (Shank et al. 2015). Hg2+ was a strong inhibitor for GPII, whereas Hg2+ interacts with sulphydeyl groups of peroxidase resulted in inhibition of enzyme activity as reported by Einollahi et al. (2006). The peroxidase of Ficus sycomorus was inhibited by Hg2+ and Cu2 (Mohamed et al. 2011a).
Conclusions
The cationic peroxidase (GPII) was purified and characterized from ginger (Zingiber officinale), which is widely used as a spice and folk medicine. The natural plant phenolic compounds as pyrogallol, catechol, and guaiacol were found to be excellent electron donors for the enzyme compared to other phenolic compounds. PGII could be used in several applications due to its catalytic properties, thermal stability, broad pH, and acting on various phenolic compounds.
Availability of data and materials
The data and materials during the current study are available in the Laboratory.
Abbreviations
- CM–Sepharose:
-
Carboxymethyl-Sepharose
- DEAE–Sepharose:
-
Diethylaminethyl-Sepharose
- H2O2 :
-
Hydrogen peroxide
- kDa:
-
Kilo Dalton
- Km :
-
Micael’s constant
- PGII:
-
Cationic peroxidase
- SDS-polyacrylamide:
-
Sodium dodecylsulphate-polyacrylamide
- Vmax :
-
Maximum velocity
References
Abdel-Aty AM, Hamed MB, Gad AM, El-Hakim AE, Mohamed SA (2018) Ficus sycomorus latex: an efficient alternative Egyptian source for horseradish peroxidase in labeling with antibodies for immunodiagnostic kits. Vet World 11:1364–1370
Abdel-Aty AM, Salama WH, Fahmy AS, Mohamed SA (2019) Impact of germination on antioxidant capacity of garden cress: new calculation for determination of total antioxidant activity. Sci Hort 246:155–160
Aghelan Z, Shariat SZS (2015) Partial purification and biochemical characterization of peroxidase from rosemary (Rosmarinus officinalis L.) leaves. Adv Biomed Res 4:159
Akhani SP, Vishwakarma SL, Golyal RK (2004) Anti-diabetic activity of Zingiber officinale in streptozotocin-induced type I diabetic rats. J Pharm Pharmacol 56:101–105
Aslmoshtaghi E, Shahsavar AR (2016) Peroxidase, polyphenol oxidase and protein changes in olives during adventitious root formation. Trakia J Sci 2:76–182
Balasubramanian M, Boopathy R (2013) Purification and characterization of peroxidases from liquid endosperm of Cocos nucifera (L.): biotransformation. J Mol Cataly B: Enzym 90:33–42
Battistuzzi G, D’Onofrio M, Loschi L, Sola M (2001) Isolation and characterization of two peroxidases from Cucumis sativus. Arch Biochem Biophys 388:100–112
Bhatti HN (2007) Kinetics of irreversible thermal denatuiration of horseradish peroxidases. J Chem Soc Pak 29:161–165
Bhatti HN, Kalsoom U, Habib A (2012) Decolorization of direct dyes using peroxidase from Raphanus sativus (F04 SL). J Chem Soc Pak 34:257–262
Bhatti HN, Najma A, Asghar M, Hanif MA, Zia MA (2006) Purification and thermal characterization of a novel peroxidase from a local chick pea cultivar. Protein Peptide Lett 13:799–804
Boucoiran CFS, Kijne JW, Recourt K (2000) Isolation and partial characterization of a thermostable isoperoxidases from potato (Solanum tuberosum L.) tuber sprouts. J Agric Food Chem 48:701–707
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Chari KLN, Manasa D, Srinivas P, Sowbhagya HB (2013) Enzyme-assisted extraction of bioactive compounds from ginger (Zingiber officinale Roscoe). Food Chem 139:509–514
Chen L-C, Chung Y-C, Chang C-T (2012) Characterisation of an acidic peroxidase from papaya (Carica papaya L. cv Tainung no. 2) latex and its application in the determination of micromolar hydrogen peroxide in milk. Food Chem 135:2529–2535
Civello PM, Arting GA, Chaves AR, Anan MC (1995) Peroxidase from strawberry fruit (Fragaria ananassa Duch. ), by partial purification and determination of some properties. J Agri Food Chem 43:2596–2601
Einollahi N, Abbasi S, Dashti N, Vaezzadeh F (2006) Effect of mercuric chloride on kinetic properties of horseradish peroxidase. Iranian J Publ Health 35:49–56
Galende PP, de María CG, Arellano JB, Roig MG, Shnyrov VL (2016) Study on extraction, purification and characterization of a novel peroxidase from white spanish broom (Cytisus Multiflorus). Int J Plant Biol Res 4:1052
Has-Schön E, Lepeduš H, Jerabek L, Cesar V (2005) Influence of storage temperature on total peroxidase activity in crude extracts from Picea abies L. Karst. needles. Croatica Chemica Acta CCACAA 78:349–353
Heinzkill M, Bech L, Halkier T, Schneider P, Anke T (1998) Characterization of laccases and peroxidases from wood-rotting fungi (family Coprinaceae). Appl Environ Microbiol 64:1601–1606
Huang CN, Horng JS, Yin MC (2004) Antioxidative and antiglycative effects of six organosulfur compounds in low-density lipoprotein and plasma. J Agric Food Chem 52:3674–3678
Joy O, Eze SOO (2015) Partial purification and characterizaton of peroxidase extracted from Gongronema latifolium. American-Eurasian J Sci Res 10:221–227
Julião MMS, Oliveira ST, Andrade LBS, Figueiredo MF, Salles HO (2016) Partial purification and thermal stability of two peroxidases from pithecellobium dulce (roxb.) benth. Aril Rev Virtual Quim 8:1913–1923
Kalsoom U, Ashraf SS, Bhatti HN (2013) Mechanistic study of a diazo dye degradation by soybean peroxidase. Chem Cent J 7:93. https://doi.org/10.1186/1752-153X-7-93
Khatun S, Ashraduzzaman M, Karim R, Pervin F, Absar N, Rosma A (2012) Purification and characterization of peroxidase from Moringa oleifera L. leaves. BioResources 7:3237–3251
Kondeti RS, Korivi M, Kesireddy N, Hua KC, Kesireddy SR (2011) Protective effect of dietary ginger on antioxidant enzymes and oxidative damage in experimental diabetic rat tissues. Food Chem 124:1436–1442
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Lavery CB, Macinnis MC, Macdonald MJ, Williams JB, Spencer CA, Burke AA, Irwin DJ, D'Cunha GB (2010) Purification of peroxidase from Horseradish (Armoracia rusticana) roots. J Agric Food Chem 58:8471–8476
Mathé C, Barre A, Jourda C, Dunand C (2010) Evolution and expression of class III peroxidases. Arch Biochem Biophys 500:58–65
Miranda MV, Fernandez Lahor HM, Cascone O (1995) Horseradish peroxidase extraction and purification by aqueous two-phase partition. Appl Biochem Biotechnol 53:147–154
Mirazizi F, Bahrami A, Haghbeen K, Zahiri HS, Bakavoli M, Legge RL (2016) Rapid and direct spectrophotometric method for kinetics studies and routine assay of peroxidase based on aniline diazo substrates. J Enzyme Inhib Med Chem 31:1162–1169
Mohamed SA, Abdel-Aty AM, Hamed MB, Mohamed OE, Fahmy AS (2011a) Ficus sycomorus latex: a thermostable peroxidase. Afr J Biotechnol 10:17532–17543
Mohamed SA, Abulnaja KO, Ads AS, Khan JA, Kumosani TA (2011b) Characterization of an anionic peroxidase from horseradish cv. Balady. Food Chem 128:725–730
Mohamed SA, Al-Harbi MH, Almulaiky YQ, Ibrahim IH, El-Shishtawy RM (2017) Immobilization of horseradish peroxidase on Fe3O4 magnetic nanoparticles. Electron J Biotechnol 27:84–90
Mohamed SA, Awad MA, Al-Qurashi AD (2014) Antioxidant activity, antioxidant compounds, antioxidant andhydrolytic enzymes activities of ‘Barhee’ dates at harvest and during storage as affected by pre-harvest spray of some growth regulators. Sci Hort 167:91–99
Mohamed SA, El-Badry MO, Drees EA, Fahmy AS (2008) Properties of a cationic peroxidase from Citrus Jambhiri cv. Adalia. Appl Biochem Biotechnol 150:127–137
Moubasher H, Mostafa FA, Wahsh S, Haroun O (2017) Purification and characterization of lignin peroxidase isozymes from Humicola grisea (Traaen) and its application in bioremediation of textile dyes. Egypt J Bot 57:335–343
Nouren S, Bhatti HN, Bhatti IA, Asgher M (2013) Kinetic and thermal characterization of peroxidase from peels of Citrus reticulata var. kinnow. J Plant Animal Sci 23:430–435
Nouren S, Bhatti HN, Ilyas S, Nazli ZI, Bibi I (2015) Citrus limon peroxidase catalyzed decolorization of textile industry effluents and toxicological evaluation of their degraded products. Fresenius Environ Bull 24:4685–4695
Osuji AC, Eze SO, Osayi EE, Chilaka FC (2014) Biobleaching of industrial important dyes with peroxidase partially purified from garlic. Sci World J 2014:183163
Padiglia A, Medda R, Cruciani E (1995) Fractionation and characterization of two form of peroxidase from Oryza sativa. Prep Biochem 25:11–19
Pandey VP, Manika A, Swati S, Sameeksha T, Dwivedi UN (2017) A comprehensive review on function and application of plant peroxidases. Biochem Anal Biochem 6:308
Pinto MST, Ribeiro JM, Francisco PA, Melo NF, Fernandes KVS (2015) Purification and characterization of a peroxidase present in xilopodium exsudates of umbu plants (Spondias tuberosa A.). Afr J Biotechnol 14:1838–1845
Ros-Barceló A, Pomar F (2002) Plant peroxidases: versatile catalysts in the synthesis of bioactive natural products. Stud Nat Prod Chem 27:735–792
Sarvamangala RP (2014) Production and purification of lignin peroxidase from Bacillus megaterium and its application in bioremediation. CIB Tech J Microbiol 3:22–28
Shank LP, Kijjanapanich P, Phutrakul S (2015) Characterization of partially purified peroxidase from finger root (Boesenbergia Rotunda (L.) Mansf.). J Med Bioeng 4:170–177
Shigeto J, Tsutsumi Y (2015) Diverse functions and reactions of class III peroxidases. New Phytol 209:1395–1402
Shukla A, Gundampati RK, Jagannadham MV (2016) Biochemical and biophysical characterization of a peroxidase isolated from Euphorbia tirucalli with antifungal activity. Biocataly Biotransform 34:236–248
Tabassum A, Bhatti HN, Nouren S, Zahid M (2015) Catalytic potential of gourd peel peroxidase for biodegradation of synthetic recalcitrant dyes fuchsin acid and crystal violet. J Plant Animal Sci 25:777–783
Tognolli M, Penel C, Greppin H, Simon P (2002) Analysis and expression of the class III peroxidase large gene family in Arabidopsis thaliana. Gene 288:129–138
Yamazaki I, Yokota K (1973) Oxidation states of peroxidase. Mol Cell Biochem 15:39–52
Yeh H, Chuang C-H, Chen H-C, Wan C-J, Chen T-L, Lin L-Y (2014) Bioactive components analysis of two various gingers (Zingiber officinale Roscoe) and antioxidant effect of ginger extracts. LWT-Food Sci Technol 55:329–334
Acknowledgements
The authors gratefully acknowledge the National Research Centre, Cairo, Egypt, for providing the facilitation and equipment throughout the study.
Funding
There is no funding.
Author information
Authors and Affiliations
Contributions
MIE-K and AMA-E participated in the purification of the enzyme. MFD, MIE-K, and AMA-E carried out the characterization of the enzyme. ASF and SAM participated in the design of the study, performed the statistical analysis, and helped to revise the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
El-Khonezy, M.I., Abd-Elaziz, A.M., Dondeti, M.F. et al. Purification and characterization of cationic peroxidase from ginger (Zingiber officinale). Bull Natl Res Cent 44, 11 (2020). https://doi.org/10.1186/s42269-019-0264-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42269-019-0264-x