
Abstract
Background
Antibiotic resistance occurs rapidly and naturally. However, the misuse of antibiotics is accelerating the process. And therefore, exploring new antibiotics has been a great demand in order to save people’s life. Actinobacteria have been the major source of antibiotics. In this study, we focused on rare types of actinobacteria which are hard to isolate from the environment by traditional methods. Fifty rare actinobacteria were isolated from Egyptian soils, and they were screened against some bacterial pathogens (Staphylococcus aureus ATCC 6538, Pseudomonas aeruginosa ATCC 10145, Klebsiella pneumonia CCM 4415, Streptococcus mutans ATCC 25175, Escherichia coli O157:H7 ATCC 51659, and Salmonella enterica ATCC 25566). Illumina whole genome sequencing was performed for potent isolates. The whole genomes of selected rare actinobacteria were investigated via bioinformatics analysis using neighbor-joining phylogenetic analysis and Antibiotics and Secondary Metabolite Analysis SHell.
Results
Isolates Rc5 and Ru87 showed the highest inhibition activity against selected Gram-positive and Gram-negative pathogens. Neighbor-joining phylogenetic analysis confirmed that isolate Rc5 belonged to Micromonospora oryzae and Micromonospora harpali with 73% bootstrap value while isolate Ru87 was grouped with Streptomyces gingianensis and Streptomyces morales with 89% bootstrap value. Bioinformatics analysis using antiSMASH 3.0 predicted 33 and 19 secondary metabolite gene clusters in Micromonospora sp. Rc5 and Streptomyces sp. Ru87, respectively. Gene annotation predicted the presence of valuable biosynthetic gene clusters in both strains such as polyketides, non-ribosomal peptides, terpenes, siderophores, bacteriocin, lasso peptide, ectoine, and lantipeptide.
Conclusion
We concluded that exploring cryptic and novel biosynthetic gene clusters via Illumina whole genome sequencing and bioinformatics analysis is a useful method. We confirmed that Egyptian soil is very rich in high potential biosynthetic of rare actinobacteria. Further genetic engineering manipulation of biosynthetic pathways would eventually lead to producing novel bioactive molecules.
Similar content being viewed by others

Introduction
In silico prediction and characterization of a microbial secondary metabolite of biosynthetic gene clusters has successfully contributed to the development of new medicines (Loureiro et al. 2018). The majority of these metabolites belong to a wide variety of chemical classes and have shown antibiotic and antitumor activities (Law et al. 2019). Recently, the cost of genome sequencing has dramatically decreased and allows for the discovery of thousands of gene clusters encoding the biosynthetic machinery for these compounds (Palazzotto and Weber 2018). The experimental description of each gene cluster is still very difficult and cannot catch up with the rapidity of genomic discovery. Therefore, effective in silico prediction of the most promising targets within the genomes is essential for the successful genomic mining (Horn et al. 2015; Foulston 2019; Sun et al. 2019).
Rare actinobacteria have been recognized as one of the great sources for bioactive compounds and antibiotic (Bredholdt et al. 2007; Khanna et al. 2011; Hacene et al. 2000; Laidi et al. 2006; Balagurunathan and Radhakrishnan 2010). Uncommon and novel types of actinobacteria may contain unexpected genes that are phylogenetically distant from related strains and, subsequently, generate alternative novel antimicrobial agents (Donadio et al. 2005).
Illumina genome sequencing has proven to be an effective approach for genomics study and sequence analysis of individual genes, clusters of genes or operons, full chromosomes, or entire genomes of any organism (Bentley 2006; Castro et al. 2018). Complete sequences of the Streptomyces coelicolor and Streptomyces avermitilis genomes and many Streptomyces genome sequences showed a lot of silent gene clusters that encode bioactive molecules (Bentley et al. 2002; Choi et al. 2015). Several years ago, the traditional fermentation process revealed that Streptomyces ambofaciens produces spiramycin and congocidine. The advancement of genome mining approach has explored many other gene clusters within rare actinobacteria of potential bioactive molecules such as kanamycin derivatives and stambomycin, yet many bioactive molecules remain hidden within silent biosynthetic gene clusters (Aigle et al. 2014; Genilloud 2018).
The advancements in bioinformatics analysis of biosynthetic gene cluster identification have facilitated the processing of enormous genomic data of actinobacteria (Alam et al. 2011; Doroghazi et al. 2014; Abdelmohsen et al. 2015; Hug et al. 2018). Manual annotation found to be difficult, inefficient, and leading to inadequate annotations, while automated annotation of secondary metabolite clusters would lead to more accurate and complete annotations. In silico genomic prediction has successfully provided great analysis of secondary metabolism in bacterial genomes, to list few examples: ClustScan (Cullum et al. 2011), SBSPKS toolbox (Anand et al. 2010), and NP.searcher web server (Li et al. 2009a). Antibiotics and Secondary Metabolite Analysis SHell (antiSMASH) has generated rapid genome annotation of a varied range of bacterial and fungal strains (Medema et al. 2011; Blin et al. 2013; Villebro et al. 2019; Hu et al. 2019).
In the present study, Illumina whole genome sequencing was conducted along with software pipeline antiSMASH.3 for the analysis and annotation of secondary metabolite gene cluster. The antiSMASH database is considered a broad resource for the secondary metabolite biosynthetic gene clusters and encompasses gene clusters for more than 3000 finished bacterial genomes (Blin et al. 2017). This eventually allowed the identification and detection of all recognized classes of the secondary metabolite biosynthetic gene clusters in our strains. The detailed functional annotation was obtained and therefore opening the door for combinatorial biosynthesis for designing novel scaffolds of antibiotics.
Material and methods
Microorganism and their maintenance
Rare actinomycete isolates were cultivated using both starch casein broth and soya bean broth as described by Abbas and Edwards (1990) with slight modification as follows: Spore suspension of the isolates was cultivated into 35 ml of each of the broth media for 7 days at 30 °C. The incubation was conducted by shaking at 150 RPM. The final spore suspension was lypholized at the Mycological center, Assiut University, Assiut, Egypt, and then kept at − 20 °C.
The antimicrobial potentialities of the 50 rare actinobacteria isolates were tested against some bacterial pathogens (methicillin-resistant Staphylococcus aureus “ATCC 6538,” Streptococcus mutans “ATCC 25175,” Escherichia coli “O157:H7 ATCC 51659,” Klebsiella pneumonia “CCM 4415,” Salmonella enterica “ATCC 25566,” and Peudomonas aeuroginosa “ATCC 10145”). Tested pathogens were provided from Ain Shams Specialized Hospital and the Microbial Resources Center (MIRCEN) at the Faculty of Agriculture, Ain Shams University, Cairo, Egypt. Cultivation of these strains was conducted overnight in nutrient broth at 37 °C.
Agar well diffusion method
Agar well diffusion method was used for preliminary screening of the antimicrobial activity of the rare actinobacteria against tested bacterial pathogens. Actinobacteria spore suspensions were prepared. Cell-free supernatant was obtained by centrifugation for 5 min at 37 °C with a speed of 100 RPM. An amount of 250 μl of cell-free supernatant was added to each well in the nutrient agar Petri dishes containing 150 μl of 0.5 McFarland of tested bacterial spores (McFarland 1907). Petri dishes were then incubated for 24 h at 37 °C. Results were recorded by measuring the inhibition zone of bacterial pathogen around the well (Cooper 1972). All tests and experiments were made in duplicates. The most potent actinobacteria Micromonospora sp. Rc5 and Streptomyces sp. Ru87 were selected for further studies (Amin et al. 2017a; Amin et al. 2017b).
Extraction of genomic DNA from actinomycete strains
Extraction of genomic DNA of Micromonospora Rc5 and Streptomyces Ru87 was conducted using Promega Wizard® Genomic DNA Purification Kit as follows: 1 ml of each actinomycete spore suspension (3 × 105 CFU/ml) was aseptically added to 35 ml of sterile starch casein broth in 50-ml Erlenmeyer flasks. The flasks were incubated at 30 °C in a shaking incubator (Spectronics, USA) with an agitation rate (150 RPM) for 7 days. One milliliter of spore suspension was added to a 1.5-ml microcentrifuge tube. Cells were pelleted by centrifugation at 13,000×g for 2 min, and the supernatant was discarded. Cells were resuspended thoroughly in a mixture of 480 μl of 50 mM EDTA (Tris-acetic acid EDTA buffer) and 120 μl of Lysozyme. Incubation of the samples was conducted at 37 °C for 60 min followed by centrifugation for 2 min at 13,000×g. The supernatant was discarded, and 600 μl of Nuclei Lysis Solution was added followed by incubation at 80 °C for 5 min with an immediate cooling down to 37 °C. Three microliters of (4 mg/ml) RNase Solution was added to the lysate. The Eppendorf tubes were then inverted five times to allow complete mixing followed by incubating at 37 °C for 60 min. An aliquot of 200 μl of protein precipitation solution was added to the RNase-treated cell lysate and vigorously vortexed at high speed for 20 s. The mixture was incubated in ice for 5 min and then centrifuged at 13,000×g for 3 min. The supernatant containing the target DNA was transferred to a clean 0.5-ml Eppendorf tube containing 600 μl of isopropanol. Eppendorf tubes were gently inverted until the thread-like strands of DNA formed a visible mass. Samples were centrifuged at 13,000×g for 2 min, and the supernatant was poured off carefully followed by draining the tubes on a clean absorbent paper. An amount of 600 μl of 70% ethanol was added to each Eppendorf tube, and tubes were then inverted several times in order to wash the DNA pellet followed by centrifugation at 13,000×g for 2 min. Careful aspiration of the ethanol was performed, and then, the tubes were poured on a clean absorbent paper to allow the pellet to air-dry for 15 min. One hundred microliters of DNA rehydration solution was added to each tube and incubated at 65 °C for 1 h. DNA concentration was determined using a Nanodrop spectrophotometer (ND-2. 1000, Nanodrop Technologies) and stored at − 20 °C.
Agar gel electrophoresis
To check the integrity and quality of the extracted DNA, an aliquot of 5 μl of each sample was loaded on 1% Agarose Gel Electrophoresis in Tris-acetic acid EDTA buffer for 30 min at 90 V. The gel was stained with 50 mg/ml of ethidium bromide, and digital images were obtained for the DNA bands of expected size using UV transilluminator (Bio-Rad Laboratories, Hercules, CA).
Illumina whole genome sequencing and contig assembly
High-quality DNA extracted from actinobacteria mycelium was sequenced using Illumina MiSeq and HiSeq 2500 platforms using 2 × 250 bp paired-end technology by Microbes NG (http://www.microbesng.uk), which is supported by the BBSRC (grant number BB/L024209/1). The bioinformatics analysis was provided by the sequencing company using Trimmomatic to trim raw reads (Bolger et al. 2014) and other software such as Samtools (Li et al. 2009b) and bwa-mem (Li and Durbin 2009) to quality filter the reads and assemble the genome. The assembly metrics provided by MicrobesNG were calculated using QUAST (Quality Assessment Tool for Genome Assemblies). The taxonomic distribution of both strains was calculated using the Kraken software (Wood and Salzberg 2014). Phylogenetic tree of complete 16S rRNA genes of isolate Rc5 and Ru87 was constructed using the neighbor-joining method (Saitou and Nei 1987). Sequence similarity was conducted using Clustal W within the Mega 7 program. Contigs obtained from the sequenced genomes of each strain were assembled on the basis of reference genomes (Micromonospora carbonaceae in case of Rc5 and Streptomyces coelicolor in case of Ru87), and gaps were filled using Mauve Aligner 2.4. software (Rissman et al. 2009). Artemis software 16.0.11 (http://www.sanger.ac.uk/Software/Artemis) was used in order to allow visualization of sequence features and assembling of contigs and plotting of whole genomes of both isolates.
Annotation of rare actinobacteria genome
Gene annotation was performed via the NCBI (National Center for Biotechnology Information) Prokaryotic Genome Annotation Pipeline (PGAAP) as published online (Tatusova et al. 2016), and an additional annotation was done using Prokka version 1.1 (Seemann 2014) to assist identifying gene clusters. In this study, we used gene annotation via an antiSMASH web server version 3.0.5 for automatic whole genomic identification and analysis of biosynthetic gene clusters in each isolate (Medema et al. 2011).
Results
Antimicrobial potential of rare actinomycete isolates
Fifty rare actinomycete isolates were selectively isolated by physical and chemical means on humic acid vitamin agar media and starch casein agar media from different Egyptian governorates. The isolates were previously identified via morphological, chemotaxonomy, and biochemical methods (Abd-allah et al. 2012). The antimicrobial activity of these isolates was screened against tested bacterial pathogens (Staphylococcus aureus ATCC 6538, Pseudomonas aeruginosa ATCC 10145, Klebsiella pneumonia CCM 4415, Streptococcus mutans ATCC 25175, Escherichia coli ATCC 51659, and Salmonella enterica ATCC 25566).
Results obtained under shaking conditions using soya bean meal broth indicated that 32 (64%) rare actinobacteria showed pronounced antimicrobial activities against the selected bacterial pathogens while it was 29 (58%) on starch casein broth medium (Table 1). In the case of using soya bean broth, 27 isolates (84%) were active against Pseudomonas aeruginosa ATCC 10145, followed by 9 (28%) against Escherichia coli ATCC 51659 and 7 (21%) in case of both Staphylococcus aureus ATCC 6538 and Salmonella enterica ATCC 25566. Four isolates (12%) were active against Streptococcus mutans 25175, while only 3 isolates (9%) were active against Klebsiella pneumonia CCM 4415. The antimicrobial activity of rare actinobacteria growing on starch casein broth reported 23 (79%), 8 (27%), and 7 (24%) active against Pseudomonas aeruginosa ATCC 10145, Staphylococcus aureus ATCC 6538, and Klebsiella pneumonia CCM 4415, respectively. Four isolates (13%) were active against Streptococcus mutans 25175, and 3 isolates (1%) showed activity against Salmonella enterica ATCC 25566. No antimicrobial activity was recorded against Escherichia coli ATCC 51659. Our data indicated that isolate number Rc5 and Ru87 had the highest antimicrobial activity. They also produced a broad-spectrum antimicrobial compound(s) against both Gram-positive and Gram-negative tested pathogenic microorganisms.
Illumina sequencing genome notification
Digital images of agarose gel captured by UV trans illuminator (Bio-Rad Laboratories, Hercules, CA) confirmed the high quality of DNA extracted from Micromonospora Rc5 and Streptomyces Ru87. The whole genome of Micromonospora sp. Rc5 contains 2252-Mb raw reads with 128.284 coverage. The assembly consists of 513 contigs. The draft genome was 7,702,789 bp, with an average GC content of 73.64%. A total of 6792 coding sequences (CDS) with 6504 coding genes were identified by NCBI prokaryote pipeline. In the case of Streptomyces sp. Ru87, the draft genome was 7,662,503 bp, with an average GC content of 73.12% was assembled in 629 contigs. NCBI prokaryote pipeline annotation identified 6527 coding sequences (CDS) with 6051 coding genes, with an average GC content of 73.12%. The taxonomic distribution of both strains calculated using the software Kraken emphasized that Micromonospora sp. Rc5 belongs to genus Micromonospora, while Streptomyces sp. Ru87 belongs to genus Streptomyces. Micromonospora sp. Rc5 Whole Genome Sequencing Bio project has been deposited at EMBL (European Molecular Biology Laboratory)/GenBank under no. PRJNA354176 (BioSample SAMN06041774, Accession MQMK00000000). Streptomyces sp. Ru87 Whole Genome Sequencing Bio project has been deposited at EMBL/GenBank under no. PRJNA413750 (BioSample SAMN07765385, Accession PDIX00000000). Micromonospora sp. Rc5 16S rRNA gene sequence was deposited in Genbank under the accession number KY818317.1 and KY818662.1 for Streptomyces sp. Ru87.
Sequence analysis and phylogenetic tree construction
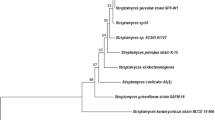
The whole genomes were sequenced via Illumina and then subjected to genome assembly. Genome annotation using NCBI prokaryote pipeline revealed 16S rRNA genes for each strain. Gene sequences were successfully deposited in Genbank under the accession number KY818317.1 for isolate Rc5 and KY818662.1 for isolate Ru87. The 16S rRNA gene sequence of isolate Rc5 was compared with other Micromonospora gene sequences in the NCBI GenBank database. The phylogenetic tree was generated against closely related Micromonospora strains using the neighbor-joining method. Neighbor-joining (NJ) phylogenetic tree consisted of two main clades. Moreover, isolate Rc5 was gathered with other Micromonospora strains in the same clade, which ensures that isolate Rc5 belonged to this genus. The most similarity of 16S rRNA gene sequence belonged to Micromonospora oryzae and Micromonospora harpali with 73% bootstrap value (Fig. 1).

Phylogenetic tree of complete 16S rRNA genes of isolate Rc5. Sequence similarity was conducted using Clustal W within the Mega 7 program. Phylogenetic trees were constructed using the neighbor-joining method
A comprehensive analysis of complete 16S rRNA Streptomyces gene tree was conducted in order to clarify the relationship between Ru87 isolate and closely related Streptomyces species. The 16S rRNA gene sequence of strain Ru87 was compared with the nucleotide sequences of other Streptomyces strains in the NCBI GenBank database. The phylogenetic tree was generated based on the comparison between the 16S rRNA gene sequence of the strain Ru87 and other nucleotide sequences from closely related Streptomyces strains. Isolate Ru87 was grouped with Streptomyces gingianensis and Streptomyces morales partial sequence with 89% bootstrap value (Fig. 2). Sequencing analysis of the 16S rRNA gene sequence confirmed that Rc5 and Ru87 were identified as Micromonospora sp. Rc5 and Streptomyces sp. Ru87.

Phylogenetic tree of complete 16S rRNA genes of isolate Ru87. Sequence similarity was conducted using Clustal W within the Mega 7 program. Phylogenetic trees were constructed using the Neighbor-Joining method
Genome mining of whole genome sequence of selected rare actinobacteria using antiSMASH analysis
Biosynthetic pathways are very important in novel antibiotic discovery. Whole genome sequences of Micromonospora sp. Rc5 and Streptomyces sp. Ru87 obtained from the Illumina sequencing were assembled on the basis of reference genomes Micromonospora carbonaceae for Rc5 and Streptomyces coelicolour for Ru87. The gaps were filled using mauve Aligner 2.4. software (Rissman et al. 2009). Genomes were mined using the antiSMASH server for further prediction of secondary metabolite biosynthetic gene clusters. The genomes sequenced of Micromonospora sp. Rc5 displayed more diverse antiSMASH readout than Streptomyces sp. Ru87. A total of 33 potential secondary metabolite gene clusters were predicted by Micromonospora sp. Rc5, 5 polyketide synthase (PKS), 4 non-ribosomal polyketide synthase (NRPS), 10 hybrid polyketide synthases, 4 terpenes, 3 lantipeptides, 2 saccharides, 1 siderophore, 1 bacteriocin, 1 arylpolyene, and 2 unidentified clusters (Table 2). Micromonospora sp. Rc5 whole genome recorded the highest similarity hits with biosynthetic gene clusters coding for the following compounds: sioxanthin (terpene) (100%), SapB (lantipeptide) (100%), desferrioxamie B (siderophore) (66%), methoxyhydroquinones (PKS) (57%), and tetrocarcin A (PKS-hybrid) (53%) (Table 3).
In the case of Streptomyces sp. Ru87, antiSMASH generated 19 potential secondary metabolite gene clusters encoding for 4 NRPSs, 4 terpenes, 2 PKS, 2 bacteriocins, 2 lasso peptide, 1 siderophore, 1 lantipeptide, 1 hserlactone, 1 ectoine, and 1 linaridin (Table 2). The whole genome of Streptomyces sp. Ru87 showed the highest similarity hits with the biosynthetic clusters coding for ectoine (100%), paenibactin (NRPS) (66%), albachelin (NRPS) (60%), erythrochellin (NRPS) (42%), and labrinthopeptin (lantipeptide) (40%) (Table 3).
Discussion
Actinobacteria generate extensive compounds with a variety of biological activities (Bredholdt et al. 2007; Khanna et al. 2011), and rare actinobacteria are known as a great potential source of antibiotic production (Hacene et al. 2000; Laidi et al. 2006; Balagurunathan and Radhakrishnan 2010). We believe that uncommon and rare types of actinobacteria may contain unexpected genes. These genes are phylogenetically far off related strains and, subsequently, generate alternative novel bioactive molecules (Donadio et al. 2005).
In this study, isolates Rc5 and Ru87 showed great inhibition activity against selected Gram-positive and Gram-negative pathogens. Illumina genome sequencing in combination with bioinformatics analysis using antiSMASH software proved to be an effective approach for genomics study and sequence analysis for the selected strains (Bentley 2006; Jakubiec-Krzesniak et al. 2018). The genome size of Micromonospora sp. Rc5 and Streptomyces sp. Ru87 was approximately similar, and it resembles the normal genome size variation among other actinobacteria which is from 4 to 12 Mb (Komaki et al. 2016; Jiang et al. 2015). Our results confirmed that Rc5 and Ru87 isolates are distinct from comparable strains on the database. Our previous phylogenetic analysis via Sanger sequencing of partial 16S rRNA genes (Amin et al. 2017a; Amin et al. 2017b) is in complete agreement with other complete 16S rRNA genes generated with Illumina whole genome sequencing. This will definitely confirm that using Sanger sequencing for characterization of partial 16S rRNA gene (V3 region) of actinobacteria is effective in genera identification. Moreover, whole genome sequencing in taxa identification is expensive and needs more data analysis (Sims et al. 2014; Luo et al. 2014). Further investigations including DNA-DNA hybridization, additional chemotaxonomic, and biochemical tests are required to identify their species level.
The antiSMASH readout of our actinobacteria is in agreement with other publications (Horn et al. 2015) and indicated a direct relation between genome size and biosynthetic potential. However, Micromonospora sp. Rc5 with larger genome size displayed more diverse (antiSMASH) read-out than Streptomyces sp. Ru87. These results showed the higher antimicrobial potential of Micromonospora sp. Rc5 than Streptomyces sp. Ru87. GC content is the main measure of the relatedness of microorganisms; it varies with different organisms due to variation in selection, mutational bias, and biased recombination-associated DNA repair (Birdsell 2002). So, we clarified that fluctuation in genome size and GC content is species dependent.
Both strains have shown great potential to produce post-translationally modified peptides such as terpenes, lantipeptides, saccharides, siderophores, bacteriocin, arylpolyene, lasso peptide, hserlactone, ectoine, and linaridins. Illumina whole genome sequencing results confirmed that both strains possess polyketides and non-ribosomal peptide gene clusters which are the major classes of pharmacologically active natural products. This ensures that the rare actinobacteria produce different metabolites synthesized by conserved enzymes (Komaki et al. 2016; Gomez-Escribano and Bibb 2012). This obviously shed the light on the high metabolic potential of the two actinomycete species to generate diverse bioactive molecules. It is of interest to mention that the combination of both genomics-metabolics sketchings of rare actinobacteria led to the characterization of cryptic or undiscovered biosynthetic clusters with a new mode of action to inhibit resistant bacteria (Foulston 2019; Xu and Wright 2019).
Actinobacteria strains Rc5 and Ru87 were tested in vitro and showed significant antibacterial activity. This antagonistic activity found to be in agreement with antiSMASH pipeline prediction of several biosynthetic gene clusters. Physicochemical analysis of the purified antimicrobial compounds was performed in order to determine the possible chemical group for each strain (Amin et al. 2017a; Amin et al. 2017b). The physicochemical analysis and bioinformatics genome mining confirmed the ability of Micromonospora sp. Rc5 to produce tetrocarcin antibiotic harboring phthalate core and caused inhibitory effect against S. aureus ATCC 6238. We have found that the labyrinthopeptin structure is in agreement with the physicochemical analysis of the active fraction produced by Streptomyces sp. Ru87, which is cyclic or aromatic peptide structure. Our data indicated that the mutual understanding from in silico and in vitro approaches leads to the identification of the closest possible antimicrobial compound produced by actinobacteria strains. This is a unique and infrequent approach of Illumina whole genome sequencing for observing the biosynthetic clusters of antibiotic-producing actinobacteria in Egypt.
Conclusion
Our findings illustrated that the Egyptian soil is very rich of high potential biosynthetic of rare actinobacteria. The genetic potential of secondary bioactive molecule producers was successfully determined via genome mining. Sequencing actinomycete genomes provide useful information for inventing novel antimicrobial agents. We ensure that rare actinobacteria genome sequencing guided with bioinformatics analysis will open the door for scientists to explore more about the biochemical pathways and consequently the discovery of novel bioactive molecules. This approach would contribute to more discovery of natural antibiotics and therefore enhance pharmaceutical industry. The current study helps to control the problem of antimicrobial drug resistance and improve the health care in Egypt, the UK, and worldwide. In addition, it introduces potential bioactive agents that would support the drug discovery in Egypt.
Abbreviations
- antiSMASH:
-
Antibiotics and Secondary Metabolite Analysis SHell
- CDS:
-
Coding sequences
- EDTA:
-
Ethylene diamine tetra acetic acid
- EMBL:
-
European Molecular Biology Laboratory
- MIRCEN:
-
Microbial Resources Center
- NCBI:
-
National Center for Biotechnology Information
- NJ phylogenetic tree:
-
Neighbor-joining phylogenetic tree
- NRPS:
-
Non-ribosomal peptide synthetase
- PGAAP:
-
Prokaryotic Genomes Automatic Annotation Pipeline
- PKS:
-
Polyketide synthase
- QUAST:
-
Quality Assessment Tool for Genome Assemblies
References
Abbas AS, Edwards C (1990) Effects of metals on Streptomyces coelicolor growth and actinorhodin production. Appl Environ Microbiol 56(3):675–680
Abd-allah N, Tolba S, Hatem D (2012) Selective isolation of rare actinomycetes from different types of Egyptian soil. Egypt J Exp Biol 8(2):175–182
Abdelmohsen UR, Grkovic T, Balasubramanian S, Kamel MS, Quinn RJ, Hentschel U (2015) Elicitation of secondary metabolism in actinomycetes. Biotechnol Adv 33(6):798–811
Aigle B, Lautru S, Spiteller D, Dickschat JS, Challis GL, Leblond P et al (2014) Genome mining of Streptomyces ambofaciens. J Ind Microbiol Biotechnol 41(2):251–263
Alam MT, Medema MH, Takano E, Breitling R (2011) Comparative genome-scale metabolic modeling of actinomycetes: the topology of essential core metabolism. FEBS Lett 585(14):2389–2394
Amin DH, Abolmaaty A, Tolba S, Abdallah NA, Wellington EM (2017a) Phylogenic characteristics of a unique antagonistic micromonospora Sp. Rc5 to S. aureus isolated from Sinai Desert of Egypt. Curr Res Microbiol Biotechnol 5(6):1295–1306
Amin DH, Tolba S, Abolmaaty A, Abdallah NA, Wellington EM (2017b) Phylogenetic and antimicrobial characteristics of a novel Streptomyces sp. Ru87 isolated from Egyptian soil. Int J Curr Microbiol App Sci 6(8):2524–2541
Anand S, Prasad M, Yadav G, Kumar N, Shehara J, Ansari MZ et al (2010) SBSPKS: structure based sequence analysis of polyketide synthases. Nucleic Acids Res 38(suppl_2):W487–WW96
Balagurunathan R, Radhakrishnan M (2010) Biotechnological, genetic engineering and nanotechnological potential of actinomycetes. Industrial exploitation of microorganisms, pp 302–436
Bentley DR (2006) Whole-genome re-sequencing. Curr Opin Genet Dev 16(6):545–552
Bentley SD, Chater KF, Cerdeno-Tarraga A-M, Challis GL, Thomson N, James KD et al (2002) Complete genome sequence of the model actinomycete Streptomyces coelicolor A3 (2). Nature. 417(6885):141–147
Birdsell JA (2002) Integrating genomics, bioinformatics, and classical genetics to study the effects of recombination on genome evolution. Mol Biol Evol 19(7):1181–1197
Blin K, Kim HU, Medema MH, Weber T (2017) Recent development of antiSMASH and other computational approaches to mine secondary metabolite biosynthetic gene clusters. Brief Bioinform. https://doi.org/10.1093/bib/bbx146
Blin K, Medema MH, Kazempour D, Fischbach MA, Breitling R, Takano E et al (2013) antiSMASH 2.0—a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res 41(W1):W204–WW12
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30(15):2114–2120
Bredholdt H, Galatenko OA, Engelhardt K, Fjærvik E, Terekhova LP, Zotchev SB (2007) Rare actinomycete bacteria from the shallow water sediments of the Trondheim fjord, Norway: isolation, diversity and biological activity. Environ Microbiol 9(11):2756–2764
Castro JF, Razmilic V, Gomez-Escribano JP, Andrews B, Asenjo J, Bibb M (2018) The ‘gifted’actinomycete Streptomyces leeuwenhoekii. Antonie Van Leeuwenhoek 111(8):1433–1448
Choi S-S, Kim H-J, Lee H-S, Kim P, Kim E-S (2015) Genome mining of rare actinomycetes and cryptic pathway awakening. Process Biochem 50(8):1184–1193
Cooper K (1972) Thetheory of antibiotic diffusion zones. Analytical Microbiology II Aca-demic Press, Inc, London, pp 13–30
Cullum J, Starcevic A, Diminic J, Zucko J, Long PF, Hranueli D (2011) ClustScan: an integrated program package for the detection and semiautomatic annotation of secondary metabolite clusters in genomic and metagenomic DNA datasets. In: Handbook of molecular microbial ecology I: metagenomics and complementary approaches, pp 423–432
Donadio S, Sosio M, Stegmann E, Weber T, Wohlleben W (2005) Comparative analysis and insights into the evolution of gene clusters for glycopeptide antibiotic biosynthesis. Mol Gen Genomics 274(1):40–50
Doroghazi JR, Albright JC, Goering AW, Ju K-S, Haines RR, Tchalukov KA et al (2014) A roadmap for natural product discovery based on large-scale genomics and metabolomics. Nat Chem Biol 10(11):963–968
Foulston L (2019) Genome mining and prospects for antibiotic discovery. Curr Opin Microbiol 51:1–8
Genilloud O (2018) Mining actinomycetes for novel antibiotics in the omics era: are we ready to exploit this new paradigm? Antibiotics. 7(4):85
Gomez-Escribano JP, Bibb MJ (2012) Streptomyces coelicolor as an expression host for heterologous gene clusters. Methods Enzymol 517:279–300
Hacene H, Daoudi-Hamdad F, Bhatnagar T, Baratti J, Lefebvre G (2000) H107, a new aminoglycoside anti-Pseudomonas antibiotic produced by a new strain of Spirillospora. Microbios. 102(402):69–77
Horn H, Cheng C, Edrada-Ebel R, Hentschel U, Abdelmohsen UR (2015) Draft genome sequences of three chemically rich actinomycetes isolated from Mediterranean sponges. Mar Genomics 24:285–287
Hu D, Gao C, Sun C, Jin T, Fan G, Mok KM et al (2019) Genome-guided and mass spectrometry investigation of natural products produced by a potential new actinobacterial strain isolated from a mangrove ecosystem in Futian, Shenzhen, China. Sci Rep 9(1):823
Hug J, Bader C, Remškar M, Cirnski K, Müller R (2018) Concepts and methods to access novel antibiotics from actinomycetes. Antibiotics. 7(2):44
Jakubiec-Krzesniak K, Rajnisz-Mateusiak A, Guspiel A, Ziemska J, Solecka J (2018) Secondary metabolites of actinomycetes and their antibacterial, antifungal and antiviral properties. Pol J Microbiol 67(3):259–272
Jiang Y, Huang Y-h, Long Z-e (2015) De novo whole-genome sequence of Micromonospora carbonacea JXNU-1 with broad-spectrum antimicrobial activity, isolated from soil samples. Genome Announc 3(2):e00174–e00115
Khanna M, Solanki R, Lal R (2011) Selective isolation of rare actinomycetes producing novel antimicrobial compounds. Int J Adv Biotechnol Res 2(3):357–375
Komaki H, Ichikawa N, Hosoyama A, Hamada M, Harunari E, Ishikawa A et al (2016) Draft genome sequence of Micromonospora sp. DSW705 and distribution of biosynthetic gene clusters for depsipeptides bearing 4-amino-2, 4-pentadienoate in actinomycetes. Stand Genomic Sci 11(1):84
Laidi RF, Kansoh AL, Elshafei A, Cheikh B (2006) Taxonomy, identification and biological activities of a novel isolate of Streptomyces tendae. Arab J Biotechnol 9(3):427–436
Law JW-F, Ser H-L, Ab Mutalib N-S, Saokaew S, Duangjai A, Khan TM et al (2019) Streptomyces monashensis sp. nov., a novel mangrove soil actinobacterium from East Malaysia with antioxidative potential. Sci Rep 9(1):3056
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 25(14):1754–1760
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N et al (2009b) The sequence alignment/map format and SAMtools. Bioinformatics. 25(16):2078–2079
Li MH, Ung PM, Zajkowski J, Garneau-Tsodikova S, Sherman DH (2009a) Automated genome mining for natural products. BMC Bioinformatics 10(1):185
Loureiro C, Medema MH, van der Oost J, Sipkema D (2018) Exploration and exploitation of the environment for novel specialized metabolites. Curr Opin Biotechnol 50:206–213
Luo C, Rodriguez-r LM, Konstantinidis KT (2014) MyTaxa: an advanced taxonomic classifier for genomic and metagenomic sequences. Nucleic Acids Res 42(8):e73–e7e
McFarland J (1907) The nephelometer: an instrument for estimating the number of bacteria in suspensions used for calculating the opsonic index and for vaccines. J Am Med Assoc 49(14):1176–1178
Medema MH, Blin K, Cimermancic P, de Jager V, Zakrzewski P, Fischbach MA et al (2011) antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res 39(suppl_2):W339–WW46
Palazzotto E, Weber T (2018) Omics and multi-omics approaches to study the biosynthesis of secondary metabolites in microorganisms. Curr Opin Microbiol 45:109–116
Rissman AI, Mau B, Biehl BS, Darling AE, Glasner JD, Perna NT (2009) Reordering contigs of draft genomes using the Mauve aligner. Bioinformatics. 25(16):2071–2073
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4(4):406–425
Seemann T (2014) Prokka: rapid prokaryotic genome annotation. Bioinformatics. 30(14):2068–2069
Sims D, Sudbery I, Ilott NE, Heger A, Ponting CP (2014) Sequencing depth and coverage: key considerations in genomic analyses. Nat Rev Genet 15(2):121
Sun C, Yang Z, Zhang C, Liu Z, He J, Liu Q et al (2019) Genome mining of Streptomyces atratus SCSIO ZH16: discovery of atratumycin and identification of its biosynthetic gene cluster. Org Lett 21:1453–1457
Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Nawrocki EP, Zaslavsky L et al (2016) NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res 44(14):6614–6624
Villebro R, Shaw S, Blin K, Weber T (2019) Sequence-based classification of type II polyketide synthase biosynthetic gene clusters for antiSMASH. J Ind Microbiol Biotechnol 46(3–4):469–475
Wood DE, Salzberg SL (2014) Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol 15(3):R46
Xu M, Wright GD (2019) Heterologous expression-facilitated natural products’ discovery in actinomycetes. J Ind Microbiol Biotechnol 46(3–4):415–431
Acknowledgements
We would like to thank the Microbial Resources Center (Cairo MIRCEN) and Ain Shams Specialized Hospital for providing the strains of food-borne and blood-borne pathogen strains.
Funding
We are very grateful for the Scholarship provided by the Egyptian missions and British council in Egypt (Newton-Mosharafa program 2016–2017) to complete and conduct the molecular studies at the School of Life Sciences, Lab C123, University of Warwick, UK.
Availability of data and materials
Not applicable.
Author information
Authors and Affiliations
Contributions
This work was carried out in collaboration between all authors. Authors NAA, ST, AA, and EMHW designed the study and wrote the protocol. Authors DHA and CB managed the lab work of the study. Author AA managed the paper organization. Authors DHA and AA wrote the first draft of the manuscript. Authors DHA and AA managed the literature searches. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Amin, D.H., Abolmaaty, A., Borsetto, C. et al. In silico genomic mining reveals unexplored bioactive potential of rare actinobacteria isolated from Egyptian soil. Bull Natl Res Cent 43, 78 (2019). https://doi.org/10.1186/s42269-019-0121-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42269-019-0121-y