
Abstract
Cerebellar liponeurocytoma is a rare benign tumor of the central nervous system affecting adults and mainly the posterior fossa. Its positive diagnosis remains difficult due to its rarity and the presence of several differential diagnoses including medulloblastoma. The oncogenetic mechanism, the therapeutic modalities and results are still currently under study. Very few cases have been published in the literature. The aim of this review is to report, through the available data, all its various clinico-epidemiological, pathological, radiological, genetic, therapeutic and evolutionary features. In fact, this tumor is associated with a slow-growing character, appears in young adulthood with slight female predominance and is often manifested by intracranial hypertension symptoms. It is pathologically characterized by a focal component of lipomatous differentiation and a low proliferation index. It is usually presented in imaging by a well-limited mass with signal attenuation for fatty tissue and heterogeneous contrast. No mutation has been identified yet. The gold standard treatment of this tumor is maximal complete resection. The evolutionary profile is marked by frequent local recurrence. Radiotherapy could be discussed in case of recurrence, incomplete surgery, inoperable cases and cases with high proliferation index, and there is to date no proof of benefit for systemic treatment. Due to the numerous similarities of this entity with medulloblastoma, it should be always evoked face to any suspicion of medulloblastoma in adults in order to avoid a wrong overtreatment.
Similar content being viewed by others

Introduction
Cerebellar liponeurocytoma is a rare benign tumor often arising from the posterior fossa, firstly described by Bechtel et al. [1] in 1978 through the case of a 44-year-old man. This tumor was initially named “lipomatous medulloblastoma” because of its better prognosis compared with classic medulloblastoma.
In 2000, the World Health Organization (WHO) referred to this entity as liponeurocytoma and classified it as a grade I benign tumor.
However, due to the recurrences reported in the literature and the discovery of certain atypical histological aspects with a few cases of anaplasia, this tumor was reclassified as Grade II by the WHO in 2007 [2].
Currently, this entity is part of the low-grade neuronal and glioneuronal tumors (Grade II) according to the 2016 WHO classification [3].
It is a very rare tumor whose frequency has not been specified from all benign tumors and all adult central nervous system (CNS) tumors.
It appears to occur in young adults, with a slight female predominance [4, 5].
Its positive diagnosis remains difficult, given its rarity and the presence of several differential diagnoses, including medulloblastoma.
A confrontation of clinical, epidemiological, radiological, pathological and sometimes evolutionary profile of this tumor is still necessary to make a certain positive diagnosis.
The oncogenetic mechanism, treatment modalities and results are still currently under study. Very few cases have been published in the literature.
The aim of this review is to describe the current data concerning this unusual entity by reporting its various clinico-epidemiological, radiological, pathological, genetic, therapeutic and evolutionary features.
Clinico-epidemiological features
Liponeurocytoma is a slow-growing benign tumor that seems to appear in young adulthood, with a slight female predominance [4].
In Fact, Oudrhiri et al. reported a mean age of onset at 49 years old [32–79 years old] with a sex ratio of 1.8/1 [4]. Similarly, Gembruch et al., in a large systemic review of liponeurocytoma, found a mean age of onset at 45 years old [4–77 years old], with a slight female predominance (1.2/1) [5].
This contrasts with the age distribution of classic medulloblastoma, which occurs in almost 80% of cases in children [6]. However, medulloblastoma may rarely occur in adults in 20% of medulloblastomas, 6% of posterior fossa tumors and 1% of brain tumors [6].
Liponeurocytoma appears mainly in the posterior fossa in 82% of the cases [5]. It affects the cerebellar hemisphere in 51% of cases, the vermis in 12%, more rarely both cerebellar hemisphere and vermis in 4% and the cerebellopontine angle in 1% [5].
A few rare cases have been reported of liponeurocytomas of the supratentorial region, which have been labeled “central liponeurocytomas” [5].
In the majority of cases, liponeurocytoma has a unique distribution, although rare cases of multifocal liponeurocytoma have been reported in the literature [7].
Clinically, in the majority of cases, liponeurocytoma is manifested by intracranial hypertension symptoms such as headache, nausea, vomiting, vertigo, ataxia and gait imbalance [8].
Pathological features
Cerebellar liponeurocytoma is histologically characterized by the presence of monomorphic small neuronal cells with neurocyte-like cells and a focal component of lipomatous differentiation that constitutes the main histological feature of this entity [9].
The monomorphic small cells extremely resemble to the small blue cells of classical medulloblastoma as much as the cells of oligodendrogliomas or the clear cells of ependymoma [9].
Anaplastic features such as mitoses, nuclear atypia, angiogenesis and areas of necrosis are rare or even absent and are seen mainly in recurrent cases with reduction or disappearance of the lipomatous component [10].
Biologically and immunohistochemically, the tumor is presented with a low proliferation index (Ki67/MIB-1) of between 1 and 3% [10].
Both cell types (monomorphic and pseudo-adipocytes) are positive for neuronal markers such as synaptophysin, neuron-specific enolase (NSE) and microtubule-associated protein 2 (MAP2). This supports the neuronal rather than adipose origin of these cells [10]. Focal expression of glial fibrillary acidic protein (GFAP) indicates glial differentiation which is found in the majority of cases [10].
According to the 2016 WHO classification, liponeurocytoma is a grade II benign neuronal and glioneuronal tumor [3].
At present, no predictive histological markers for a high recurrence potential have yet been identified. However, studies of the pathological appearance of recurrences have shown histological factors of more aggressive growth, such as high proliferation index, vascular proliferation and tumor necrosis [11].
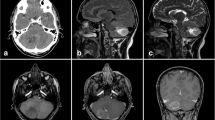
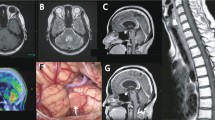
Radiological features
The radiological aspect of the tumor is variable, but may be suggestive of the diagnosis.
The scannographic appearance is most often based on a well-limited mass with signal attenuation for fatty tissue and heterogeneous contrast. The tumor may be associated with parenchymal cysts or cerebellar hemorrhage [12].
On magnetic resonance imaging (MRI), the tumor usually appears as hypo-T1, relatively well limited, with numerous hyper-T1 foci corresponding to lipid zones. Gadolinium contrast enhancement is moderate and heterogeneous [12].
Genetic characteristics
A familial predisposition has been suggested by some authors, with a possible autosomal dominant mode of inheritance [13]. However, no genetic mutation causing liponeurocytoma has yet been established.
Genetic analysis by Horstmann et al. [3] found mutations in the tumor protein53 (TP53) gene in 20% of cases. However, no mutations were found in the protein patched homolog 1 (PTCH1), adenomatous polyposis coli (APC) or beta-catenin genes (markers of medulloblastoma subtypes). In addition, they noted the non-expression of the 17q iso-chromosome classically found in medulloblastoma [14].
Cluster analysis of DNA expression profiles revealed a similarity between cerebellar liponeurocytoma and neurocytomas. However, TP53 mutations were absent in central neurocytomas which led to explore other recent markers such as neurogenin-1 (NEUROG1) and fatty acid-binding protein 4 (FABP4) [8, 15]. The results of these studies demonstrated an over expression of FABP4. This led to the conclusion that liponeurocytoma could be the consequence of the transformation of the progenitor cells of the cerebellum into adipose tumor cells through possible aberrant differentiation [8, 15].
Therapeutic and evolutionary characteristics
Maximal complete resection is the treatment of choice for liponeurocytomas [16, 17]. However, the recurrence rate remains high, estimated as 60% at a mean follow-up of 6.5 years [1–12 years] [3].
On the other hand, distant metastasis appears to be exceptional. In fact, an intra-medullary lumbar metastasis has been reported in a single case, 11 years after the initial diagnosis [18].
Surgical revision remains the first-line treatment at the recurrence time [19].
Given the low proliferation index (< 5%) reported by the majority of authors, the benefit of radiotherapy has been debated for these tumors with a slow evolutionary profile [20] and the benefit of adjuvant chemotherapy has not yet been proven [10].
According to Gembruch et al., immediate postoperative radiotherapy was carried out in 24% of cases, at a dose of 54 Gy with conventional fractionation (1.8–2 Gy per fraction) [5].
This improved local disease prognosis by 36.5% and the recurrence rate was 8.33% and 44.83% for adjuvant radiotherapy and surgery alone, respectively [5].
However, as there are currently no solid arguments in favor of systematic adjuvant radiotherapy, its indication should be discussed on a case-by-case basis at the multidisciplinary consultation meeting face to recurrence, incomplete surgery, inoperable cases or cases with a high proliferation index [5, 10].
Conclusion
Cerebellar liponeurocytoma is a rare neuro-oncological entity that remains little known to date. Its diagnosis should always be evoked face to any suspicion of medulloblastoma in adults, given their great similarity. Hence, a clinical, epidemiological, radiological and pathological confrontation must always be carried out in order to avoid diagnostic errors. The treatment of choice remains based on a maximal complete resection of the tumor. Postoperative radiotherapy appears to reduce the risk of tumor recurrence and should be proposed in cases of incomplete resection or inoperable tumor. Further studies are needed in the future to better understand this rare entity and determine its best therapeutic management.
Availability of data and materials
All used studies in this review are mentioned in the References heading.
Abbreviations
- APC:
-
Adenomatous polyposis coli
- FABP-4:
-
Fatty acid-binding protein 4
- GFAP:
-
Glial fibrillary acidic protein
- MAP2:
-
Microtubule-associated protein 2
- MRI:
-
Magnetic resonance imaging
- NEUROG1:
-
Neurogenin-1
- NSE:
-
Neuro-specific enolase
- PTCH1:
-
Protein patched homolog 1
- TP53:
-
Tumor protein53
- WHO:
-
World Health Organization
References
Bechtel JT, Patton JM, Takei Y. Mixed mesenchymal and neuroectodermal tumor of the cerebellum. Acta Neuropathol. 1978;41(3):261–3. https://doi.org/10.1007/BF00690447. (PMID: 206094).
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114(2):97–109. https://doi.org/10.1007/s00401-007-0243-4. (Epub 2007 Jul 6. Erratum in: Acta Neuropathol. 2007 Nov;114(5):547. PMID: 17618441; PMCID: PMC1929165).
Horstmann S, Perry A, Reifenberger G, Giangaspero F, Huang H, Hara A, et al. Genetic and expression profiles of cerebellar liponeurocytomas. Brain Pathol. 2004;14(3):281–9. https://doi.org/10.1111/j.1750-3639.2004.tb00065.x.
Oudrhiri MY, Raouzi N, El Kacemi I, El Fatemi N, Gana R, Maaqili MR, et al. Understanding cerebellar liponeurocytomas: case report and literature review. Case Rep Neurol Med. 2014;2014: 186826. https://doi.org/10.1155/2014/186826.
Gembruch O, Junker A, Mönninghoff C, Ahmadipour Y, Darkwah Oppong M, Sure U, et al. Liponeurocytoma: systematic review of a rare entity. World Neurosurg. 2018;120:214–33. https://doi.org/10.1016/j.wneu.2018.09.001.
Giordana MT, Schiffer P, Lanotte M, Girardi P, Chio A. Epidemiology of adult medulloblastoma. Int J Cancer. 1999;80(5):689–92. https://doi.org/10.1002/(sici)1097-0215(19990301)80:5%3c689::aid-ijc10%3e3.0.co;2-g.
Scoppetta TL, Brito MC, Prado JL, Scoppetta LC. Multifocal cerebellar liponeurocytoma. Neurology. 2015;85(21):1912. https://doi.org/10.1212/WNL.0000000000002156.
Chakraborti S, Mahadevan A, Govindan A, Yasha TC, Santosh V, Kovoor JM, et al. Supratentorial and cerebellar liponeurocytomas: report of four cases with review of literature. J Neurooncol. 2011;103(1):121–7. https://doi.org/10.1007/s11060-010-0361-z.
Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131(6):803–20. https://doi.org/10.1007/s00401-016-1545-1.
Soffietti R, Duffau H, Bauman G, Walker D. Neuronal and mixed neuronal–glial tumours. In: Batchelor T, Nishikawa R, Tarbell N, Weller M, editors. Oxford Textbooks in Clinical Neurology. Oxford Textbook of Neuro-Oncology. Oxford: Oxford Academic; 2017. https://doi.org/10.1093/med/9780199651870.003.0008.
Radke J, Gehlhaar C, Lenze D, Capper D, Bock A, Heppner FL, et al. The evolution of the anaplastic cerebellar liponeurocytoma: case report and review of the literature. Clin Neuropathol. 2015;34(1):19–25. https://doi.org/10.5414/NP300783.
Alkadhi H, Keller M, Brandner S, Yonekawa Y, Kollias SS. Neuroimaging of cerebellar liponeurocytoma: case report. J Neurosurg. 2001;95(2):324–31. https://doi.org/10.3171/jns.2001.95.2.0324.
Wolf A, Alghefari H, Krivosheya D, Staudt MD, Bowden G, Macdonald DR, et al. Cerebellar liponeurocytoma: a rare intracranial tumor with possible familial predisposition: case report. J Neurosurg. 2016;125(1):57–61. https://doi.org/10.3171/2015.6.JNS142965.
Zurawel RH, Allen C, Chiappa S, Cato W, Biegel J, Cogen P, et al. Analysis of PTCH/SMO/SHH pathway genes in medulloblastoma. Genes Chromosomes Cancer. 2000;27(1):44–51. https://doi.org/10.1002/(sici)1098-2264(200001)27:1%3c44::aid-gcc6%3e3.0.co;2-v.
Anghileri E, Eoli M, Paterra R, Ferroli P, Pollo B, Cuccarini V, et al. FABP4 is a candidate marker of cerebellar liponeurocytomas. J Neurooncol. 2012;108(3):513–9. https://doi.org/10.1007/s11060-012-0853-0.
Abuzneid YS, Alzeerelhouseini HIA, Shkokani S, Aqel W, Aldarawish A. Cerebellar liponeurocytoma, a rare tumor: case report and review of the literature. Int J Surg Case Rep. 2021;82: 105937. https://doi.org/10.1016/j.ijscr.2021.105937.
Broggi G, Tirrò E, Alzoubi H, Arcella A, Gianno F, Antonelli M, et al. Cerebellar liponeurocytoma: clinical, histopathological and molecular features of a series of three cases, including one recurrent tumor. Neuropathology. 2022;42(3):169–80. https://doi.org/10.1111/neup.12799.
Soylemezoglu F, Soffer D, Onol B, Schwechheimer K, Kleihues P. Lipomatous medulloblastoma in adults: a distinct clinicopathological entity. Am J Surg Pathol. 1996;20(4):413–8. https://doi.org/10.1097/00000478-199604000-00003.
Xu N, Cai J, Du J, Yang R, Zhu H, Gao P, et al. Clinical features and prognosis for intraventricular liponeurocytoma. Oncotarget. 2017;8(37):62641–7. https://doi.org/10.18632/oncotarget.16024.
Châtillon CE, Guiot MC, Roberge D, Leblanc R. Cerebellar liponeurocytoma with high proliferation index: treatment options. Can J Neurol Sci. 2009;36(5):658–61. https://doi.org/10.1017/s0317167100008210.
Acknowledgements
Not applicable.
Funding
There is no funding to declare in this review.
Author information
Authors and Affiliations
Contributions
AJ collected data and wrote the manuscript, FD was supervising the work, WS, NF and JD read and corrected the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jribi, A., Dhouib, F., Siala, W. et al. Cerebellar liponeurocytoma: clinico-epidemiological, pathological, radiological, genetic, therapeutic and evolutionary characteristics—a review of the literature. Egypt J Neurosurg 39, 39 (2024). https://doi.org/10.1186/s41984-024-00304-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41984-024-00304-6