
Abstract
Background
Plasmacytomas of the skull base are not commonly encountered in clinical practice, with few reported cases in the literature. They form part of the spectrum of plasma cell neoplasms and are classified as a solitary bone plasmacytoma if arising from the sphenoid bone. Its radiographic appearance can lead to misdiagnosis as one of the tumours that are more frequently seen in the skull base, especially meningiomas. Due to the risk of evolution into multiple myeloma, accurate diagnosis is essential.
Case presentation
A 56-year-old male presented to the emergency department with rapid proptosis and worsening vision in his right eye for one week’s duration. Imaging studies revealed an extra-axial right sphenoid bone tumour with invasion into the temporalis muscle and orbit, leading to significant proptosis. Tumour debulking was done, but there was no improvement in vision postoperatively. Final histology was consistent with a plasmacytoma. The patient was referred to the oncologist for radiation therapy, but subsequently developed further lesions consistent with multiple myeloma.
Conclusions
Plasmacytomas need to be considered in the differential diagnosis of skull base tumours. Due to their excellent response to radiation, these patients should have early oncology intervention to prevent irreversible neurological deficits.
Similar content being viewed by others


Introduction
Plasmacytomas of the skull base are very rarely seen in clinical practice, and the literature contains only few case reports over several decades. They form part of the spectrum of plasma cell neoplasms and are classified as solitary bone plasmacytoma (SBP) or extramedullary plasmacytoma (EMP). Those that arise from the sphenoid bone are SBP. The radiographic appearance can result in its misdiagnosis as a more commonly seen skull base tumour, for example a meningioma. These tumours can potentially develop into multiple myeloma (MM). We present a rare case of this skull base tumour that caused rapidly progressive proptosis and subsequent visual loss, highlighting the fact that this tumour should be considered as an uncommon cause of proptosis. We briefly discuss our management of this patient and perform a literature review of the few documented cases.
Case description
A 56-year-old male presented to the emergency department with a one-week history of progressive visual loss in his right eye. He noted that the right eye appeared larger than the left. He denied any headaches, tinnitus, vomiting, seizures, fever or symptoms in the contralateral eye. He had no constitutional symptoms of weight loss, change in appetite or malaise.
Physical examination revealed orbital proptosis and chemosis of the right eye, but no bruit was auscultated. He had complete ophthalmoplegia of the right eye, and vision was reduced to finger counting only. The remainder of the cranial nerves were intact, and the rest of the examination unremarkable.
Computed tomography (CT) scan of the brain showed a mildly hyperdense mass originating from the right medial sphenoid wing and eroding the bones of the skull base. There was extension superiorly to the temporalis fossa and into the muscle, and antero-laterally into the orbital cavity thus causing proptosis (Fig. 1). CT imaging of the chest, abdomen and pelvis was performed to rule out a primary metastatic process, which was negative. Hematological studies including complete blood count, renal and liver function, calcium, lactate dehydrogenase, alkaline phosphatase and blood film were all within normal range. Bone marrow aspirate was within normal parameters and a skeletal survey did not detect additional lesions. Serum and urine markers for gammopathy were unfortunately not available at our institute.

Preoperative CT scan of the brain and skull base. CT brain axial views, pre (A) and post (B) contrast administration, revealing right skull base tumour arising from region of sphenoid wing, and extending into orbit and temporalis muscle. Axial bone window (C) showing degree of bony erosion of temporal bone and skull base
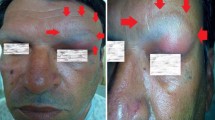
Magnetic resonance imaging (MRI) of the brain revealed a 6 cm (antero-posterior) × 5.6 cm (medio-lateral) × 7 cm (cranio-caudal) mass which appeared to arise from the medial sphenoid wing. There was no invasion of the dura, and the mass was hypointense on T1 weighted imaging. It vividly enhanced post-gadolinium administration and dural tails were noted. The mass was isointense on T2-weighted imaging, with hyperintensities centrally (Fig. 2). The differential diagnoses included skull base sarcoma, metastasis, meningioma, and plasma cell tumour. Given the rapid growth of the tumour, a hematological malignancy seemed likely. The patient’s proptosis drastically worsened (Fig. 3) over the duration over his investigations (2-week period), and a decision was made to debulk the tumour urgently. Ideally, we would perform an intraoperative consult in the form of frozen section, but this was unavailable.

Preoperative MRI brain. Top row—T1 weighted axial MRI sequences, pre (A) and post (B) gadolinium, showing degree of invasion into orbit, but lack of intra-axial spread. Evidence of a dural tail is noted on the post-contrast sequence. Bottom row—T2 coronal (C) and T1 sagittal (D) images further highlighting degree of orbital invasion by tumour

Top row—admission pictures of patient’s right eye, showing severity of proptosis. Bottom row—one-week post admission, revealing rapidity of tumour growth and worsening of proptosis
Consent was obtained for a right pterional craniotomy, with potential for orbital osteotomy. The patient was positioned supine, with the head turned 30 degrees and 3-point pinion fixation performed with the Mayfield skull clamp. A curvilinear incision was made 1 cm anterior to the tragus and extended to the midline. A myocutaneous flap was elevated and tumour was noted to be extending into the temporalis muscle and involving the bone. There was significant necrosis of the tumour centrally. Frontal and temporal bur holes were placed and a craniectomy completed. The dura was noted to be covered with tumour, but no breach was detected. The consistency of the mass was soft and easily suckable, in keeping with the appearance on T2-weighted imaging. Contiguous extension into the orbit was noted, and this tumour debulked. A gross total resection was performed, including resection of the affected temporalis muscle and bone, and a titanium cranioplasty reconstruction done (Fig. 4).

Top row—intraoperative images showing exposure of temporalis (A) and tumour overlying dura and invading temporalis muscle and temporal bone (B). Bottom row—post-resection cavity (C) and skull reconstruction with titanium mesh (D)
The patient had a rapid recovery postoperatively, but the right eye was deemed unsalvageable, and eventually enucleated by the ophthalmology team. The final histological diagnosis revealed sheets of tumour cells with plasmacytoid morphology (Fig. 5). They stained strongly for CD138 and displayed kappa light chain restriction. This was consistent with a plasma cell neoplasm. Given the origin of the tumour from the sphenoid wing and lack of additional lesions, it was classified as a solitary bone plasmacytoma.

Histological slides showing diffuse sheets of tumour cells (A) and round eccentric nuclei with abundant eosinophilic cytoplasm (B), consistent with plasmacytoid neoplasm. Picture C shows cytoplasmic and membranous staining for CD138, confirming plasma cells. There was restriction to Kappa light chain staining (D), but not to Lambda (E). This confirms a neoplastic plasma cell process
The patient was referred to the hematologic-oncology team for advice on radiotherapy. However, follow-up imaging revealed lesions in the right posterior 10th rib and clivus, in keeping with progression to MM. A decision was made to start chemo-radiotherapy, and he has not developed any further lesions at 3-month follow-up.
Discussion
Introduction
Plasmacytomas are defined as solitary or discrete masses of neoplastic plasma cells, which occur in either bone marrow or soft tissue [1]. It is part of the spectrum of plasma cell neoplasms, which includes monoclonal gammopathy of undetermined significance, solitary bone plasmacytoma, extramedullary plasmacytoma and multiple myeloma. Genetic changes that occur throughout the plasma cell lineage result in oncogenic activation and tumour suppressor cell inactivation. These include hyperdiploidy and translocation of certain loci onto oncogenes, with Cyclin-D and immunoglobulin-heavy chain dysregulation. Deletion of chromosome 13q, amplification of chromosome 1q, and deletion of chromosome 1p are secondary genetic events that also contribute to germinal centre changes, and eventual plasma cell neoplasms [2].
SBP accounts for less than 10% of plasma cell neoplasms, is more common in males with a median age of 55 years, and typically seen in vertebrae [1, 3].
Plasmacytomas of the skull base are extremely rare lesions, confined to case reports in the existing literature. SBP in this region arise from the medullary space of the sphenoid bone, clivus and petrous apex, which are rich in marrow [3]. These are classified as central skull base plasmacytomas. Those arising from the nasopharyngeal region are classified as anterior plasmacytomas and are of the EMP type [3, 4].
Clinical presentation
This is determined by the location in the skull base. Na’ara et al. [5] in 2015 reported on the signs and symptoms recorded in their analysis of these tumours. Sphenoclival tumours present with diplopia, hemianopia, blurred vision and ocular pain. Petrous apex tumours caused hearing loss vertigo and instability, orbital tumours led to proptosis, ptosis and epiphora. This is due to the proximity of particular cranial nerves in those regions.
Diagnosis
The main focus is to differentiate SBP from MM. The “CRAB” acronym for increased calcium, renal failure, anaemia and bony lesions is used to diagnose MM. SBP diagnosis requires a solitary lesion after performing skeletal survey or CT imaging, plasma cell infiltration on histologic review and normal bone marrow biopsy (less than 10% plasma cells) [6].
Imaging features include osteolytic changes to the affected bone on CT, and an extra-axial lesion that homogenously enhances post-contrast administration. T1- and T2-weighted MRI imaging reveals an isointense lesion that enhances post-gadolinium administration. There may be significant flow-voids, and even intensities along the dura resembling a dural tail. Hence, meningiomas are a major differential diagnosis, followed by hemangiopericytoma [7].
The intraoperative consultation, in the form of frozen section, has shown high diagnostic yield in cases of central nervous system (CNS) tumours. Modi et al. [8] reported its accuracy to be 92%, with a sensitivity of 93.4%. Although specific data on the use of frozen section in cases of CNS plasmacytoma is not available, this modality should be considered in the intraoperative strategy for such cases. This is due to the radiosensitive nature of these tumours, and it can guide the surgeon on the degree of resection required.
Treatment
The primary treatment is radiotherapy [7, 9, 10], with provisions for chemotherapy if the patient develops MM. Local control rates exceed 80% with radiation alone [6] and doses over 40 Grays can provide an initial response rate of over 90% [1, 3]. Surgical excision is an option if gross total resection is deemed feasible, with adjuvant radiotherapy, although its role in R0 resection is questioned [3, 5, 11].
Prognosis
There has been confirmed potential for progression to MM in both SBP and EMP, and this conversion is higher in SBP (63.6%) when compared to EMP (9.5%) [3]. At 10 years, SBP has a 65–84% progression to MM, and 65–100% at 15 years; the median time to progression is 2–3 years. Lesion size greater than 5 cm, age over 40 years, high M protein levels and persistence of M protein after treatment are thought to be risk factors for SBP progression to MM [6]. The 2-year and 5-year overall survival for skull base plasmacytomas were 78% and 59%, respectively, in an analysis by Na’ara et al. [5].
Conclusions
SBP needs to be considered as a differential diagnosis in aggressive skull base tumours, particularly in middle-aged males. While imaging features may mimic meningioma, lytic lesions of the bone are commonly seen with SBP. The primary management is radiotherapy due to its high radiosensitivity. Investigations to rule out multiple myeloma are necessary to determine both the correct treatment modality and whether a limited biopsy or surgery is preferable. Progression of SBP to MM appears to be inevitable, and patients require lifelong surveillance.
Availability of data and material
Data sharing is not applicable to this article as no datasets were generated or analysed during the current study.
Abbreviations
- SBP:
-
Solitary bone plasmacytoma
- MM:
-
Multiple myeloma
- EMP:
-
Extramedullary plasmacytoma
References
Nolan KD, Mone MC, Nelson EW. Plasma cell neoplasms: review of disease progression and report of a new variant. Surg Oncol. 2005;14(2):85–90. https://doi.org/10.1016/j.suronc.2005.05.001.
Barwick BG, Gupta VA, Vertino PM, Boise LH. Cell of origin and genetic alterations in the pathogenesis of multiple myeloma. Front Immunol. 2019;10(MAY):1121.
Wein RO, Popat SR, Doerr TD, Dutcher PO. Plasma cell tumors of the skull base: four case reports and literature review. Skull Base. 2002;12(2):77–86. https://doi.org/10.1055/s-2002-31570-1.
Vengalathur R, Kavindapadi K, Chandramouli B. Primary plasmacytoma of the anterior skull base: a rare case. Neurol India. 2014;62(5):543–6. https://doi.org/10.4103/0028-3886.144457.
Na’ara S, Amit M, Gil Z, Billan S. Plasmacytoma of the skull base: a meta-analysis. J Neurol Surg Part B Skull Base. 2015;77(1):61–5.
Kilciksiz S, Karakoyun-Celik O, Agaoglu FY, Haydaroglu A. A review for solitary plasmacytoma of bone and extramedullary plasmacytoma. Sci World J. 2012;2012:895765.
Meyer JR, Roychowdhury S, Cybulski G, Russell EJ. Solitary intramedullary plasmacytoma of the skull base mimicking aggressive meningioma. Skull Base Surg. 1997;7(2):101–5.
Modi M, Nilkanthe R, Trivedi P, Shah A, Shah J, Chaudhari J. Importance of intraoperative consultation for the diagnosis of central nervous system lesions and evaluation of its diagnostic accuracy. J Cancer Res Ther. 2018;14(6):1176–9.
Dang S, Manzoor NF, Harmsen H, Rivas A, Aulino JM. Skull base and orbital solitary plasmacytoma mimicking a meningioma. JAMA Otolaryngol Surg Publ. 2019. https://doi.org/10.1001/jamaoto.2019.2797.
Siyag A, Soni TP, Gupta AK, Sharma LM, Jakhotia N, Sharma S. Plasmacytoma of the skull-base: a rare tumor. Cureus. 2018;10(1):1–7.
Tanaka M, Shibui S, Nomura K, Nakanishi Y. Solitary plasmacytoma of the skull vault: a rare case report. Jpn J Clin Oncol. 1998;66(1):66–9.
Acknowledgements
We acknowledge the Pathology and Ophthalmology Departments of San Fernando General Hospital for their contributions to this patient’s care. Particularly, we give thanks to Dr Dawn Meyers (Pathology) for providing the histological images and an explanation of the features, which are included in Fig. 5.
Funding
No funding was received for this report.
Author information
Authors and Affiliations
Contributions
PSM and NR performed the procedure, obtained consent for publication and conceptualized the manuscript. PSM, PK and NR drafted and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Informed consent was obtained by the patient for publication of this case.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Seetahal-Maraj, P., Knight, P. & Ramnarine, N. Proptosis secondary to a solitary plasmacytoma of the sphenoid bone: a case report on a rare skull base tumour. Egypt J Neurosurg 37, 4 (2022). https://doi.org/10.1186/s41984-022-00144-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41984-022-00144-2