
Abstract
Background
Kleeblattschädel skull, also known as the cloverleaf is a complex synostosis which presents with an enlarged tri-lobar skull resulting temporal bulging and a flat posterior skull, a rare find in the population. This pansynostosis is associated with other congenital syndromes which include Crouzon, Pfeifer, and Carpenter’s. The genetic disorder Pfeiffer syndrome results from skull bones’ premature fusion characterized by deformities of the hand and feet. The author reports a 10-month-old girl who presented with a tri-lobar skull and wide toes which is a significant finding of Pfeiffer syndrome.
Case presentation
A 10-month-old girl presents with congenital obstructive hydrocephalus due to cloverleaf skull with horizontally enlarged head and large fontanelles. The child also had mid-facial hypoplasia and pre-axial bilateral lower limb polydactyly. Wide toes were also observed, an indicator of Pfeiffer’s Syndrome. Computed tomography (CAT) scans grossly abnormal craniofacial appearances, with premature closure of the sagittal suture giving a cloverleaf skull appearance. There was also a significant thinning of the brain parenchyma.
Conclusion
Premature closure of sutures leads to a forced growth in a plane perpendicular to the closed suture. Cloverleaf deformity can present with multiple syndromes such as Pfeiffer’s. This leads to gross alteration of the skull with potential underlying morbidity.
Similar content being viewed by others

Introduction
Kleeblattschädel syndrome is a complex synostosis which presents with an enlarged tri-lobar skull, enlarged fontanelles resulting in temporal bulging, and a flat posterior skull caused by the premature closure of several sutures [1]. This condition is associated with a variety of congenital abnormalities including Crouzon, Pfeiffer, and Carpenter’s. Synostosis of the sagittal and squamosal sutures may force cerebral matter through the anterior fontanelle as is in our case. Associated conditions include mid-facial hypoplasia, increased risk of increased intracranial pressure, and hydrocephalus [2]. Pfeiffer can present as wide and deviated thumbs or toes [3]. According to WHO Special Education on Pfeiffer syndrome, this birth anomaly affects 1 in 100,000 births in Western population and even rarer in Asian population of which exact data is not available.
Case presentation
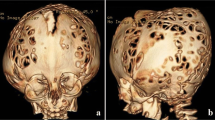
A 10-month-old girl presented as a first visit to our institute on June 2018 with a cloverleaf deformity (Fig. 1a, b), pre-axial bilateral lower limb polydactyly, mid-facial hypoplasia, exophthalmos, broad digits, and large thumb. Some milestones were delayed like inability to hold her neck, but she was smiling and had a strong hand grip. According to her attendants, she could move her head and limbs without much issue. Her feeding history included top-feed and took Cerelac on and off. There were also complains of aspiration during feed. Birth history revealed that there was increased amniotic fluid 2 days before delivery and she was in distress because of inability to take feed. Her first sibling was a still birth due to oligohydramnios.

a Patient with tri-lobar skull. b A side angle of the tri-lobar skull
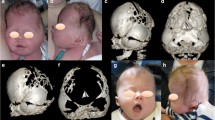
A computed tomography (CAT) scan of the head and neck was ordered which revealed premature closure of sagittal suture giving cloverleaf skull appearance with horizontally enlarged head. Bony spiculations were seen protruding between the cerebral sulci posteriorly. Gross biventricular hydrocephalus was identified, with disproportionate ballooning of the temporal horns (Fig. 2). Dilatation of the third ventricle was seen with the fourth ventricle appearing collapsed. The scan revealed a calvarial thinning with few areas of dural protrusion showing cerebrospinal fluid (CSF) density which may represent pseudo-encephalocele.

CAT showing gross biventricular hydrocephalus with disproportionate ballooning of the temporal horns
Small posterior fossa was observed with marked cerebellar tonsillar herniation up to the C2/C3 junction. Non-ossification of multiple areas in the skull was also identified (hence the incomplete 3D reconstruction images) (Fig. 3). Significant thinning of the brain parenchyma was noticed; however, there was no evidence of acute intracranial hemorrhage.

3D reconstruction imaging of the skull
The child was discharged against medical advice on request of the attendants. She was however recommended to take paracetamol, Augmentin, and lactose-free milk. The patient however was lost to follow-up.
Discussion
Classic Pfeiffer syndrome is designated as type I. Type 1 is characterized by bicoronal craniosynostosis, variable finger and toe malformations, and, in most cases, normal intellectual development. Type 2 consists of cloverleaf skull with Pfeiffer hands and feet together with ankylosis of the elbows. Type 3 is similar to type 2 but without cloverleaf skull. Ocular proptosis is severe in degree and the anterior cranial base is markedly short. Patients presenting with type 2 and type 3 usually do not do well and suffer from an early death [4].
Fearon et al. conducted a retrospective study of 802 patients treated with craniosynostosis over the past 17 years in their center. Out of the sample they tested, 28 were identified with Pfeiffer’s syndrome out of which 17 were classified as type 1 (61%), 7 as type 2 (25%), and 4 as type 3 (16%). The occurrence of hydrocephalus associated with this syndrome was 61%, and they required an intervention to treat the condition [5]. The increased intracranial pressure is manifested as bulging in the areas of the sagittal suture and squamosal sutures with a downward displacement of the ears, severe proptosis or exophthalmos, and facial deformities similar to those of craniofacial dysostosis [4]. The authors identified a 7% mortality rate associated with the syndrome and suggested a reduction in mortality rates with aggressive airway management and more frequent screening for Chiari malformations [5].
A rather unique study was done by Witt et al. who screened an 18-gestational-week fetus using standard radiographs and high-resolution CAT scans. The authors reported changes in the calvarium as being the primary focus of abnormal events leading to synostosis. This thought was opposed to the previously established concept that the synostosis was due to cranial base deformities which secondarily distorted the developing brain. This claim was inconsistent with earlier theories regarding the development of craniodysostoses, making it unique [6].
Craniosynostosis has an overall incidence of 1 in 2100 to 1 in 2500 live births with multi-suture craniosynostosis constituting about 5% of all the cases [2]. This condition is associated with a variety of presentations including optic atrophy, hydrocephalus, respiratory problems, cleft palate, and disorders of hearing and speech [7]. Pfeiffer, Apert, Crouzon, Beare–Stevenson, Jackson–Weiss, and Muenke syndromes are all associated with gain-of-function mutations in the fibroblast growth factor receptor (FGFR) 1, 2, and 3 genes [2].
According to Machado et al., the diagnosis of cloverleaf skull is purely phenotypical with observation of bitemporal bulging [8]. Cohen’s characteristic radiological findings of cloverleaf skull are a tri-lobar skull with distorted calvaria, giving a honeycomb appearance and hypoplasia of orbits along with paranasal sinuses [9].
The reconstruction of this tri-lobar skull can be done in two in two ways. The first is an early cranial vault remodeling, and the second way is a phased reconstruction, whereby a ventriculoperitoneal shunt is placed in the neonate, fronto-orbital advancement made at 3 to 6 months and posterior vault remodeling done at 1 year of age. The phased reconstruction has shown to have lesser complications in terms of pneumonia, meningitis, excessive bleeding, wound infection, seizure, and a shorter intensive care unit and hospital stay as compared to children operated for early cranial vault remodeling [10].
This deformation could lead to hydrocephalus, hindbrain herniation, and venous hypertension [9]. Our patient presented with obstructive congenital hydrocephalus and tonsillar herniation.
Conclusion
Premature closure of sutures leads to a forced growth in a plane perpendicular to the closed suture. Cloverleaf deformity can present with multiple syndromes such as Pfeiffer’s. This leads to gross alteration of the skull with potential underlying morbidity. Such presentations are rare to find in the society and should be reported for the medical community to build effective solutions to deal with the deformity. An expert team of craniofacial, maxillofacial, plastic, and neurosurgeons is needed to handle such complex conditions which involve the correction of the bony deformity, while ensuring that the brain and other soft tissues are not compromised.
Availability of data and materials
The data was obtained from the patient’s surgical records and the picture obtained with the consent of the patient.
Abbreviations
- 3D:
-
Three-dimensional
- C2/C3 junction:
-
Junction of second and third cervical vertebrae
- CAT scan:
-
Computed tomography scan
- CSF:
-
Cerebrospinal fluid
- FGFR:
-
Fibroblast growth factor receptor
References
Alvarez-Manassero D, Manassero-Morales G. Cloverleaf skull and bilateral facial clefts. Rev Chil Pediatr. 2015;86(5):357–60.
Kimonis V, Gold JA, Hoffman TL, Panchal J, Boyadjiev SA. Genetics of craniosynostosis. In Seminars in pediatric neurology 2007 (Vol. 14, no. 3, pp. 150-161). WB Saunders.
Vogels A. Fryns J. Orphanet J Rare Dis. 2006;1(1):19.
Ko JM. Genetic syndromes associated with craniosynostosis. J Korean Neurosurg Soc. 2016 May;59(3):187.
Fearon J, Rhodes J. Pfeiffer syndrome: a treatment evaluation. Plast Reconstr Surg. 2009;123(5):1560–9.
Witt P, Hardesty R, Zuppan C, Rouse G, Hasso A, Boyne P. Fetal Kleeblattschädel cranium: morphologic, radiographic, and histologic analysis. Cleft Palate-Craniofacial J. 1992;29(4):363–8.
Kirmi O, Lo SJ, Johnson D, Anslow P. Craniosynostosis: a radiological and surgical perspective. In Seminars in ultrasound, CT and MRI 2009(Vol. 30, no. 6, pp. 492-512). WB Saunders.
Machado G, Di Rocco F, Sainte-Rose C, Meyer P, Marchac D, Macquet-Nouvion G, et al. Cloverleaf skull deformity and hydrocephalus. Childs Nerv Syst. 2011;27(10):1683.
Manjila S, Chim H, Eisele S, Chowdhry SA, Gosain AK, Cohen AR. History of the Kleeblattschädel deformity: origin of concepts and evolution of management in the past 50 years. Neurosurg Focus. 2010;29(6):E7.
Jarrahy R, Kawamoto HK, Keagle J, Dickinson BP, Katchikian HV, Bradley JP. Three tenets for staged correction of Kleeblattschädel or cloverleaf skull deformity. Plast Reconstr Surg. 2009;123(1):310–8.
Funding
No funding was obtained for this study.
Author information
Authors and Affiliations
Contributions
MIS contributed to the manuscript writing. NM also contributed to the manuscript writing. SSB contributed to the manuscript review and editing. GJ contributed to the conception of idea. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The ethics approval has been taken by the Ethics and Review Committee of The Aga Khan University, Karachi, Pakistan, and a copy will be made available on the request of the editor of the journal.
Consent for publication
Consent has been sought by the patient, and the patient consent form will be made available on the request of the editor of the journal.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sajid, M.I., Malik, N., Balouch, S.S. et al. Kleeblattschädel skull presenting in concert with Pfeiffer syndrome. Egypt J Neurosurg 34, 41 (2019). https://doi.org/10.1186/s41984-019-0068-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41984-019-0068-1