
Abstract
Background
Gangliogliomas are rare tumors of the central nervous system. They can occur anywhere in the central nervous system but are most commonly located in the temporal lobe and are mainly found in children. Anaplastic ganglioglioma can result from either de novo or transformation of a pre-existing lesion.
Case presentation
We report a case of de novo anaplastic ganglioglioma in the parieto occipital region, which is a rare location. A 34-year-old lady presented with features of raised intracranial pressure (ICP) with right side hemiparesis. Contrast-enhanced magnetic resonance imaging (CEMRI) of the brain showed well-defined intense heterogenously enhancing solid cystic mass lesion 5.3 × 5.2 cm in the left parieto occipital region with mass effect and midline shift. Intraoperatively, a cystic mass lesion with reddish brown nodule was seen in the left occipital lobe. Complete tumor excision was done. Microscopic and IHC examination was suggestive of anaplastic ganglioglioma. The post-operative period was uneventful. The patient received 60-Gy radiotherapy with temozolamide as adjuvant therapy, and repeat imaging showed no tumor recurrence.
Conclusion
Anaplastic gangliogliomas are rare tumors with parieto occipital as rare location.
Similar content being viewed by others


Background
Gangliogliomas are rare tumors, and they account for 1% (reported range of 0.4 to 7%) of all central nervous system neoplasms [1]. They are commonly seen in children and young adults with males affected more commonly than females [1]. These tumors occur throughout the CNS, but the temporal lobe is the commonest site [2]. The most common presentation is seizures. They are generally of WHO grades 1 and 2, and grade 3 (anaplastic ganglioglioma) is very rare. In 5% of cases, gangliogliomas show aggressive behavior and anaplastic histopathologic features characteristic of WHO grade 3 tumors [3]. These tumors can occur de novo or as a result of malignant transformation of a pre-existing lesion. We report here a rare case of de novo occipital anaplastic ganglioglioma. On searching the English literature, a total of 41 cases of de novo anaplastic ganglioglioma have been reported with only 4 cases in parieto occipital location.
The details of the previously reported cases have been summarized in Table 1.
Case presentation
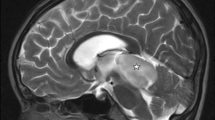
A 34-year-old female presented with complaints of on/off headache with vomiting for 1 year, which aggrevated for the last 5 days. Systemic examination was within normal limits. GCS was E4V5M6, pupils were bilateral normal size and reacting, on fundus examination-bilateral papilledema was present. On the motor system examination, right side hemiparesis was present (motor power 4/5). Contrast-enhanced magnetic resonance imaging (CEMRI) of the brain showed well-defined intense heterogenously enhancing solid cystic mass lesion 5.3 × 5.2 cm in the left parieto occipital region. Solid component was isointense on T2/FLAIR images with peripheral cystic areas and isointense on T1W images. The cystic component was iso- to hyperintense on T2/FLAIR images and hypointense on T1W images. The lesion showed mass effect in form of compression and displacement of the occipital horn of the lateral ventricle and compression of the brain stem with midline shift and brain edema. These features were suggestive of hemangioblastoma/pilocytic astrocytoma (Fig. 1).

CEMRI of the brain shows a well-defined solid cystic lesion in the left parieto occipital region. Solid component isointense and cystic component hypointense on T1 axial images (a). Solid component isointense and cystic component iso- to hyperintense on T2 axial images. Solid component shows intense enhancement on T1 contrast coronal and sagittal images (c, d), Solid component isointense and cystic component iso- to hyperintense on T2 axial images (b)
Left parieto occipital craniotomy was done which showed a cystic mass lesion with reddish brown solid nodule in the occipital region. Cyst was tapped followed by nodule excision. The nodule was soft to firm in consistency and was mild to moderately vascular. Tumor was extending into the occipital horn and midline up to the falx; however, no attachment to the falx was seen gross total tumor excision was done.
Histopathology
Microscopic examination showed cellular glial tumor composed of malignant astrocytic cells and scattered ganglion cells. Astrocytic cells showed moderate nuclear pleomorphism with coarse chromatin with few mitosis. Immunohistochemistry (IHC) analysis showed focal positivity (ganglion cells) for CD56 and was negative for CD34, P53, IDH1, and synaptophysin. MIB1 index was 8% (Figs. 2 and 3). These features were suggestive of anaplastic ganglioglioma, WHO grade 3.

Photomicrograph showing a branching capillaries and eosinophilic granular bodies seen in a fibrillary glial background. H&E, × 100. b Ganglion cells with vesicular chromatin, prominent nucleoli, and moderate to abundant amount of eosinophilic cytoplasm. H&E, × 400. c Photomicrograph showing a mitotic figure. H&E, × 100. d Cellular glial tumor composed of malignant astrocytic cells with scattered ganglion cells and marked vascular proliferation. H&E, × 100

HC a ganglion cells showing cytoplasmic positivity for CD 56. b Ki67/MIB1positivity
The immediate post-operative period was uneventful. The patient made gradual recovery over the and received 60-Gy radiotherapy with temozolamide as adjuvant therapy. Post-operative CEMRI of the brain showed craniotomy defect with gliosis in the left parieto occipital region with no contrast enhancement (Fig. 4). CEMRI of the brain was done 14 and 24 months after surgery which showed post radiotherapy changes with gliosis with no recurrent lesion (Fig. 5). The patient is asymptomatic 26 months after surgery.

Post-op CEMRI of the brain shows post-operative changes with no contrast enhancement. T1 and T2 axial (a, b) and T1 with contrast coronal and sagittal (c, d)

CEMRI brain T1 contrast image done after 14 months of surgery showing post radiotherapy changes with no enhancement
Discussion
Ganglioglioma is an infrequent tumor of the central nervous system, initially characterized by Perkins in 1926. The age of presentation of these tumors varies from 2 months to 70 years. The majority of gangliogliomas occur in the temporal lobe (> 70%) [7]. The most common presenting signs and symptoms are seizures (temporal lobe and other supratentorial locations), followed by headache, dizziness, ataxia (posterior fossa), and progressive weakness (spinal cord). The typical of ganglioglioma is a benign, calcified tumor in the temporal lobe of a child with seizures [3]. On CT, the picture is of a circumscribed solid mass or cyst with a mural nodule. On MR imaging, gangliogliomas are isointense to hypointense on T1-weighted images, are hyperintense and heterogeneous on T2-weighted images, and can contain solid, cystic, and calcified components. Enhancement after administration of gadolinium has also been found to vary from no enhancement to marked, heterogeneous enhancement [2, 12].
Histopathologically, gangliogliomas are benign, well-differentiated neuroepithelial tumors. The typical feature is a combination of neuronal and glial cell elements, which may exhibit marked heterogeneity [13]. On IHC, the glial component is positive for GFAP, S-100 protein, and vimentin, whereas the neuronal component is reactive for synaptophysin, MAP 2, NeuN, and neurofilaments [14, 15]. Histopathologic differential diagnoses comprise both high-grade and low-grade neoplasms, such as diffuse astrocytomas, oligodendrogliomas, dysembryoplastic neuroepithelial tumors, pilocytic astrocytomas (PAs), and pleomorphic xanthoastrocytomas (PXAs) [7]. Mitotic figures in ganglioglioma are rare, and MIB index varies from 1.1 to 2.7%. CD 34 is present 70–80% in the neuronal component of ganglioglioma but is less common in anaplastic variants [15]. IDH1 and P53 should be done to rule out diffuse glioma.
Anaplastic gangliogliomas typically demonstrate malignant transformation of the glial component with hypercellularity, vascular proliferation, and necrosis high mitotic labeling indices (Ki-67) [16,17,18]. Anaplastic transformation is more common in the pediatric population and has been associated with previous subtotal tumor resection and radiotherapy [19]. Gross total resection should be attempted whenever possible with the preservation of neurological function. Some studies have advocated the role of gross total resection along with adjuvant radiochemotherapy in achieving good survival rates [20, 21].
Conclusion
Anaplastic gangliogliomas are rare tumors with parieto occipital as rare location. Imaging may vary, and histology along with IHC is needed for the diagnosis. Gross total resection along with adjuvant therapy improves outcome.
Availability of data and materials
Please contact Dr. Vikas Sharma for data requests. All identifiable data (name of patients, birthdate) have been removed out of the figure scans.
Abbreviations
- CEMRI:
-
Contrast-enhanced magnetic resonance imaging
- ICP:
-
Intracranial pressure
- IHC:
-
Immunohistochemistry
- PAs:
-
Pilocytic astrocytomas
- PXAs:
-
Pleomorphic xanthoastrocytomas
References
Zhang D, Henning TD, Zou LG, Hu LB, Wen L, Feng XY, Dai SH, Wang WX, Sun QR, Zhang ZG. Intracranial ganglioglioma: clinicopathological and MRI findings in 16 patients. Clin Radiol. 2008;63(1):80–91.
Zentner J, Wolf HK, Ostertun B, Hufnagel A, Campos MG, Solymosi L, Schramm J. Gangliogliomas: clinical, radiological, and histopathological findings in 51 patients. J Neurol Neurosurg Psychiatry. 1994;57(12):1497–502.
Rumana CS, Valadka AB. Radiation therapy and malignant degeneration of benign supratentorial gangliogliomas. Neurosurgery. 1998;42:1038–43.
Demarchi R, Abu-Abed S, Munoz D, Loch Macdonald R. Malignant ganglioglioma: case report and review of literature. J Neurooncol. 2011;101:311–8.
Hirose T, Kannuki S, Nishida K, Matsumoto K, Sano T, Hizawa K. Anaplastic ganglioglioma of the brain stem demonstrating active neurosecretory features of neoplastic neuronal cells. Acta Neuropathol. 1992;83:365–70.
Wolf HK, Muller MB, Spanle M, Zentner J, Schramm J, Wiestler OD. Ganglioglioma: a detailed histopathological and immunohistochemical analysis of 61 cases. Acta Neuropathol. 1994;88:166–73.
Majores M, von Lehe M, Fassunke J, Schramm J, Becker AJ, Simon M. Tumor recurrence and malignant progression of gangliogliomas. Cancer. 2008;113:3355–63.
Romero-Rojas AE, Diaz-Perez JA, Chinchilla-Olaya SI, Amaro D, Lozano-Castillo A, Ligia I. Restrepo-Escobar histopathological and immunohistochemical profile in anaplastic gangliogliomas. neurocirugia. 2013;24:237–43.
Lucas JT Jr, Huang AJ, Mott RT, Lesser GJ, Tatter SB, Chan MD. Anaplastic ganglioglioma: a report of three cases and review of the literature. J Neuro-oncol. 2015;123:171–7.
Pikis S, Petrosyan T, Diamantopoulou K, Kelesis C. Anaplastic ganglioglioma becoming symptomatic in the third trimester of pregnancy. Int J Reprod Contracept Obstet Gynecol. 2017;6:1158–60.
Lundar T, Due-Tønnessen BJ, Fric R, Egge A, Krossnes B, Due-Tønnessen P, Stensvold E, Brandal P. Acta Neurochir (Wien). 2018;160(6):1207–14.
Johannsson JH, Rekate HL, Roessmann U. Gangliogliomas: pathological and clinical correlation. J Neurosurg. 1981;54:58–63.
Hirose T, Scheithauer BW, Lopes MB, et al. Ganglioglioma: an ultrastructural and immunohistochemical study. Cancer. 1997;79:989–1003.
Blumcke I, Giencke K, Wardelmann E, Beyenburg S, Kral T, Sarioglu N, Pietsch T, Wolf HK, Schramm J, Elger CE, Wiestler OD. The CD34 epitope is expressed in neoplastic and malformative lesions associated with chronic, focal epilepsies. Acta Neuropathol (Berl). 1999;1997:481–90.
Blumcke I, Muller S, Buslei R, Riederer BM, Wiestler OD. Microtubule-associated protein-2 immunoreactivity: a useful tool in the differential diagnosis of low-grade neuroepithelial tumors. Acta Neuropathol. 2004;108:89–96.
Kawataki T, Sato E, Sato T, Kinouchi H. Anaplastic ganglioglioma with malignant features in both neuronal and glial components-case report. Neurol Med Chir. 2010;50:228–31.
Karabekir HS, Balci C, Tokyol C. Primary spinal anaplastic ganglioglioma. Pediatr Neurosurg. 2006;42:374–8.
Nakajima M, Kidooka M, Nakasu S. Anaplastic ganglioglioma with dissemination to the spinal cord: a case report. Surg Neurol. 1998;49:445–8.
Selvanathan SK, Hammouche S, Salminen HJ, Jenkinson MD. Outcome and prognostic features in anaplastic ganglioglioma: analysis of cases from the SEER database. J Neurooncol. 2001;105:539–45.
Karremann M, Pietsch T, Janssen G, Kramm CM, Wolff JE. Anaplastic ganglioglioma in children. J Neurooncol. 2009;92:157–63.
Terrier, L. M., Bauchet, L., Rigau, V., Amelot, A., Zouaoui, S., Filipiak, I., … Club de Neuro-Oncologie of the Société Française de Neurochirurgie . Natural course and prognosis of anaplastic gangliogliomas: a multicenter retrospective study of 43 cases from the French Brain Tumor Database. Neuro-Oncol,19(5): 678–688.,2016
Acknowledgements
Not applicable.
Funding
There is no external funding for the publication of this article.
Author information
Authors and Affiliations
Contributions
VS and SB contributed to the data collection and writing of the manuscript. AH contributed to the histopathology and IHC analysis part. VS and SRH contributed to the cross-checking and editing of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
As this is a case report, ethical clearance is not required.
Consent for publication
Consent for publication was taken from the patient prior to reporting of the case.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sharma, V., Bhaskar, S., Hire, S.R. et al. A case report of rare location of ganglioglioma. Egypt J Neurosurg 34, 35 (2019). https://doi.org/10.1186/s41984-019-0060-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41984-019-0060-9