
Abstract
Background
Primary central nervous system lymphoma (PCNSL) is an uncommon extranodal non-Hodgkin lymphoma. This tumor is common in the fifth to seventh decade and is located commonly in the periventricular region and in the cerebral hemisphere. Corpus callosum is a rare site. We report a case of diffuse large B cell lymphoma with corpus callosum and contralateral side involvement in immunocompetent patient.
Case presentation
A 50-year-old male was admitted with complaints of headache with one episode of generalized seizure. On examination, the patient was neurologically intact. CEMRI (contrast-enhanced magnetic resonance imaging) brain showed heterogenous enhancing mass lesion in the left frontal and basifrontal region with involvement of rostrum and body of corpus callosum with patchy areas of diffusion restriction and choline peak on MRS (magnetic resonance spectroscopy). Intraoperatively yellowish brown tumor was seen in the left basifrontal and frontal region with involvement of corpus callosum and extension to opposite side. Microscopic and IHC (immunohistochemistry) examination was suggestive of diffuse large cell lymphoma. Postoperative period was uneventful. The patient received chemoradiotherapy in postoperative period. The patient is asymptomatic 6 months after surgery.
Conclusion
Primary CNS lymphoma is a rare tumor, and corpus callosum with contralateral side involvement is rare.
Similar content being viewed by others


Background
Primary diffuse large cell lymphomas of CNS are rare tumors, accounting for < 1% of all lymphoma and 4–6% of all extranodal lymphomas [1]. These tumors were first described by Bailey in 1929 (Intracranial sarcomatous tumors of leptomeningeal origin) as “perithelial sarcoma” of the CNS till the lymphoid lineage was clarified. Immunodeficiency is a risk factor for these tumors. By location, most common site is frontal lobe and periventricular region. Corpus callosum involvement is rare. We report a case of diffuse large B cell lymphoma in immunocompetent patient with corpus callosum involvement with extension to contralateral side.
Case presentation
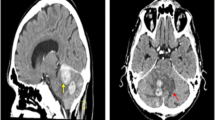
A 50-year-old male was admitted with complaints of headache for 3 months and one episode of seizure. On examination, GCS was E4V5M6 and pupils were bilaterally equal and reacting to light. No motor deficit was present. CEMRI brain showed mass lesion in the left frontal and basifrontal region with involvement of rostrum and body of corpus callosum with mass effect and compression of frontal horn of the left lateral ventricle. On T1W images, the lesion was hypo- to isointense, and on T2W images, the lesion was iso- to hyperintense. Heterogenous contrast enhancement was present with patchy areas of diffusion restriction (Fig. 1). On MRS, there was choline peak with reduced NAA (N-acetyl aspartate) peak. There was increased choline/NAA and choline/creatinine ratio (Fig. 2). The differentials were glioblastoma multiforme and CNS lymphoma. HIV was negative.

CEMRI brain shows an ill-defined mass lesion in the left frontal and basifrontal region with corpus callosum extension. Lesion is iso to hypointense on T1W image (a) and iso to hyperintense on T2W image (b) and shows heterogenous contrast enhancement (c) with patchy areas of diffusion restriction (d)

MRS shows choline peak with decreased NAA peak
The patient was operated, and left frontal lobectomy with tumor excision was done. Intraoperatively yellowish brown tumor was seen in the left basifrontal and frontal region with involvement of corpus callosum. Tumor was mildly vascular and was firm in consistency. Tumor involving corpus callosum was left behind. Subtotal excision was done (Fig. 3).

a Intra op picture after tumor removal showing the falx and frontal base. b Tumor tissue. c Post op CT Image
Histopathology-Microscopic examination showed large round to oval cells present in sheets with a high nuclear/cytoplasmic ratio. Brisk mitosis was seen. IHC (immunohistochemistry) examination was positive for LCA, CD20, CD79a and was negative for GFAP. (Figure 4) Ki-67 index was 80–85%. These features were suggestive of diffuse large B cell lymphoma. Postoperative period was uneventful. The patient received methotrexate-based chemotherapy along with radiotherapy in postoperative period and is asymptomatic 6 months after surgery.

a Photomicrograph showing large round to oval cells present in sheets with a high nuclear/cytoplasmic ratio, H&E × 40. b IHC showing CD20 positivity
Discussion
Primary central nervous system lymphoma (PCNSL) is a rare form of extranodal non-Hodgkin’s lymphoma and occurs in both immunocompromised and immunocompetent patient. The percentage of primary CNS lymphoma is 2.4–3% in all brain tumors and 4–6% of all extranodal lymphoma [1]. These tumors can occur in any age group and have a peak incidence between the fifth and seventh decade of life. However, increased incidence has been reported in age group above 60 years in the last two decades. The median patient age is 56 years, and the male-to-female ratio is 3:2.
About 60% of all PCNSLs involve the supratentorial space with the frontal lobe, basal ganglia, and periventricular region commonly involved. Corpus callosum involvement is rare. The posterior fossa and spinal cord are less frequently affected [2]. In immunocompetent patients, 90% PCNSL are diffuse large B cell (DLBCL) type and the remaining 10% are low-grade lymphomas, Burkitt’s lymphomas, or T cell lymphomas [3]. HIV-related PCNSL is typically a large cell lymphoma with immunoblastic and more aggressive features [4]. The presentations of PCNSL in both immunocompromised and immunocompetent patients is similar, with signs of a focal mass lesion in 70% and 61.3% of patients, respectively [5]. Seizures are less common than with other types of brain tumors probably because PCNSL involves predominantly subcortical white matter rather than epileptogenic gray matter. PCNSL patients do not present with B symptoms such as fever, weight loss, or night sweats.
In immunocompetent PCNSL patients, lesions are solitary in 65% of cases and are located in the cerebral hemisphere (38%), thalamus or basal ganglia (16%), corpus callosum (14%), periventricular region (12%), and cerebellum (9%) [6]. HIV-related PCNSL is solitary in 48.6% of cases and is localized to the cerebral cortex in 65%, the periventricular region in 56%, the basal ganglia in 33%, the cerebellum in 7%, and the brainstem in 4% [5].
CEMRI brain is the imaging of choice. These tumors are hypointense on T1-weighted images, isointense to hyperintense on T2-weighted images, and have homogenous dense enhancement on postcontrast images. Peritumoral edema is relatively limited and is less severe than in malignant gliomas and metastasis [7]. In HIV-related PCNSL ring enhancement is often seen [8]. Steroids, radiation and anti-cancer therapy can alter MRI findings and diagnosis may be difficult [9]. The differential diagnosis of PCNSL includes central nervous system gliomas, metastatic tumors, demyelinating disorders, subacute infarcts, and space-occupying lesions due to an infectious etiology.
On histopathological examination, PCNSLs are highly cellular, diffusely growing tumors with no specific pattern. Centrally large areas of geographical necrosis are common and may harbor viable perivascular lymphoma islands. At the periphery an angiocentric infiltration pattern is frequent. The tumor cells are mature B cells with a PAX5-positive, CD19-positive, CD20-positive, CD22-positive, and CD79a-positive phenotypes [10]. Mitotic activity is high and Ki-67 proliferation index usually exceeds 70% or even 90% [11].
Treatment involves surgical excision followed by radiochemotherapy. Gross-total resection of the tumor is typically not possible because of the infiltrative nature of the tumor [12]. High-dose methotrexate-based polychemotherapy is given [7]. Whole brain irradiation may improve outcome, but has the risk of neurotoxicity in elderly patients. Age (> 65 years) is a major negative prognostic factor and is associated with reduced survival [7, 13]. PCNSL has a poor outcome than does systemic DLBCL.
Conclusion
Primary CNS lymphomas are rare tumors. Most common type is diffuse large B cell lymphoma. The frontal lobe and periventricular region is the most common site. Corpus callosum is a rare site. CEMRI brain is the imaging of choice. Treatment involves surgical excision followed by chemoradiotherapy.
Abbreviations
- CEMRI:
-
Contrast enhanced magnetic resonance imaging
- DLBCL:
-
Diffuse large B cell lymphoma
- IHC:
-
Immunohistochemistry
- MRS:
-
Magnetic Resonance Spectroscopy
- NAA:
-
N-Acetyl aspartate
- PCNSL:
-
Primary central nervous system lymphoma
References
Schlegel U. Primary CNS lymphoma. Ther Adv Neurol Disord. 2009;2(2):93–104.
Deckert M, Engert A, Bruck W, Ferreri AJ, Finke J, Lllerhaus G, et al. Modern concepts in the biology, diagnosis, differential diagnosis and treatment of primary central nervous system lymphoma. Leukemia. 2011;25(12):1797–807.
Miller DC, Hochberg FH, Harris NL, et al. Pathology with clinical correlations of primary central nervous system non-Hodgkin’s lymphoma.The Massachusetts General Hospital experience 1958-1989. Cancer. 1994;74:1383–97.
Meeker TC, Shiramizu B, Kaplan L, et al. Evidence for molecular subtypes of HIV-associated lymphoma: division into peripheral monoclonal, polyclonal and central nervous system lymphoma. AIDS. 1991;5:669–74.
Newell M, Hoy J, Cooper S, et al. Human immunodeficiency virus-related primary central nervous system lymphoma: factors influencing survival in 111 patients. Cancer. 2004;100:2627–34.
Kuker W, Nagele T, Korfel A, et al. Primary central nervous system lymphomas (PCNSL): MRI features at presentation in 100 patients. J Neuro-Oncol. 2005;72:169–77.
Korfel A, Schlegel U. Diagnosis and treatment of primary CNS lymphoma. Nat Rev Neurol. 2013;9(6):317–27.
Fine HA, Mayer RJ. Primary central nervous system lymphoma. Ann Intern Med. 1993;119:1093–104.
Nabavizadeh SA, Vossough A, Hajmomenian M, Assadsangabi R, Mohan S. Neuro imaging in central nervous system lymphoma. Hematol Oncol Clin North Am. 2016;30(4):799–821.
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, editors. WHO classification of tumours of the central nervous system, vol. 4. Lyon: IARC; 2016. p. 272–3.
Brunn A, Nagel I, Montesinos-Rongen M, Klapper W, Vater I, Paulus W, et al. Frequent triple-hit expression of MYC, BCL2, and BCL6 in primary lymphoma of the central nervous system and absence of a favorable MYC(low)BCL2 (low) subgroup may underlie the inferior prognosis as compared to systemic diffuse large B cell lymphomas. Acta Neuropathol. 2013;126(4):603–5.
Bellinzona M, Roser F, Ostertag H, et al. Surgical removal of primary central nervous system lymphomas (PCNSL) presenting as space occupying lesions: a series of 33 cases. Eur J Surg Oncol. 2005;31:100–5.
Abrey LE, DeAngelis LM, Yahalom J. Long-term survival in primary CNS lymphoma. J Clin Oncol. 1998;16(3):859–63.
Acknowledgements
Not applicable.
Funding
There is no external funding for publication of this article.
Availability of data and materials
Please contact Dr. Vikas Sharma for data requests.
All identifiable data (name of patients, birthdate) have been removed out of the figures scans.
Author information
Authors and Affiliations
Contributions
VS and SD contributed to the data collection and writing of the manuscript. RA contributed to the histopathology and IHC analysis part. VS and SD contributed to the cross-checking and editing of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Authors’ information
Dr.Vikas Sharma is associate consultant and Dr. Sanjeev Dua is head of neurosurgery Department at Max hospital Delhi. Dr. Rooma Ambastha is pathologist in the same hospital.
Ethics approval and consent to participate
As this is a case report, this was discussed with the ethical committee members and they concluded that ethical clearance is not required for case reports.
Consent for publication
Consent for publication was taken from the patient prior to reporting of case.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sharma, V., Dua, S. & Ambastha, R. An interesting case of primary diffuse large B cell lymphoma of the central nervous system: a case report. Egypt J Neurosurg 34, 11 (2019). https://doi.org/10.1186/s41984-019-0034-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41984-019-0034-y