
Abstract
Background
Brain iron accumulation neurodegeneration (NBIA) comprises a group of rare diseases characterized by deposits of this metal in brain structures. It presents a great variability of progression, which can be fast enough to lead the patient to death in the first years of life, or slow enough to be confused with non-progressive diseases.
Case presentation
Female, 19-year-old patient, cesarean delivery at 38 weeks of gestation. History of multiple sclerosis in a second-degree paternal aunt. Normal neuropsychomotor growth and development up to 11 months, when changes in gait began. After that, it got worse, with decreased muscle strength and falls, but it progressed so slowly that she was diagnosed with cerebral palsy, a non-progressive condition. At the age of 16, she underwent molecular analysis by exome sequencing, being diagnosed with the NBIA Phospholipase A2 (PLAN) variant. Currently doing physical therapy, hydrotherapy, occupational therapy and sertraline.
Conclusions
The report draws attention to the variability of the disease and the possibility of confusion with other diagnoses, which may delay proper management.
Similar content being viewed by others


Background
Neurodegeneration with brain iron accumulation (NBIA) represents a group of neurodegenerative diseases whose main characteristic is the abnormal accumulation of iron in the basal nuclei of the brain [1, 2].
Over the years, several genetic and phenotypic mutations have been discovered and, as a result, different cases of neurodegeneration have been reported. It is a heterogeneous group of diseases with diverse clinical presentations, whose diagnosis is challenging, especially due to the possible semiological overlap with some cases of non-progressive chronic encephalopathies [2]. There are several nosological entities that have in common the deposition of iron in the basal ganglia, which can be evidenced by the classic “tiger’s eye sign” on magnetic resonance imaging [3].
The course of the disease is highly variable. Onset can occur from childhood to adulthood. Progression can be faster or slower, with long periods of stability [4]. Therefore, due to the variability of the clinical syndromes and the evident overlap between them, reaching a specific diagnosis is often challenging.
Case presentation
This case was unique due to its presentation, initially without symptoms, later presenting an extremely slow progression, being confused with a non-progressive disease leading to a late diagnosis at 16 years of age.
Female, 19 years, student, was seen at the outpatient clinic with a complaint of speech disorders. History of cesarean delivery, at 38 weeks of gestation, developing fever, needing to be hospitalized for 15 days due to neonatal infection. Her parents are not consanguineous. The patient is the first of three children. There is a family history of multiple sclerosis in a second-degree paternal aunt. The family reported that the patient had a normal growth and neuropsychomotor development; and a qualitative change in gait at 11 months. Her learning was normal, with age-appropriate schooling.
The gait was altered, since she learned to walk, receiving an initial diagnosis of spastic diplegia, with partial improvement with rehabilitation. At the age of 6, she began to suffer frequent falls, in addition to tremors and occasional numbness in her limbs in the morning. the neurological exam revealed normal speech, global hyperreflexia, mild spasticity, without motor deficit or coordination alteration. The patient was diagnosed with cerebral palsy (CP).
At the age of 8, returned to the neuropediatrician with improvement in the episodes of falls and stabilization of the condition; however, school difficulties began. She had a magnetic resonance imaging of the skull, lumbar and cervical spine with no alterations, and screening for inborn errors of metabolism was then carried out, with no evidence of abnormalities.
The clinical picture remained stable until the age of 12, when there was a worsening of the spasticity in the lower limbs, with the need for a surgical procedure due to shortening of the tendons. From this age onwards, there was a worsening of symptoms with worsening of speech, writing and motor command, in addition to an increase in episodes of falling and worsening of motor skills.
At age 16, the physical examination showed a grade 4 muscle strength in the upper limbs and a grade 3 on the lower limbs, with distal atrophy, bilateral, axial and appendicular ataxia, with hyperreflexia.
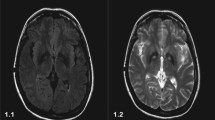
Electroneuromyography showed severe motor and sensory axonal polyneuropathy predominantly affecting the lower limbs with ongoing signs of denervation. Nuclear magnetic resonance using a MR BRIVO 355 GE—USA MRI machine revealed significant cerebellar atrophy (Fig. 1), in addition to hypointensity in the globus pallidus and substantia nigra of the midbrain bilaterally, on T2 and FLAIR sequences (Fig. 2). Molecular analysis by exome sequencing showed two heterozygous pathogenic variants in the PLA2G6 gene, consistent with a diagnosis of NBIA variant phospholipase A2 (PLAN). In the following months, there was a rapid worsening of speech and gait, requiring the use of a walker. The balance was impaired, showing Romberg's sign when closed the eyes.

MRI images of the patient’s brain, showing diffuse cerebellar atrophy on T2 sequence: A coronal. B Axial

Axial MRI images of the patient's brain showing signs of iron deposition in structures of the extrapyramidal system: A hyposignal in the substantia nigra of the midbrain in the SWI sequence. B Hyposignal in globus pallidus bilaterally in SWI sequence. C Hyposignal in globus pallidus bilaterally on DWI sequence
The patient is currently undergoing rehabilitation with physiotherapy, hydrotherapy, occupational therapy and drug therapy (using sertraline for mood swings). The younger sister started showing the same symptoms at age 7 and is under investigation.
Early diagnosis of cerebral palsy is still an unreached goal in most underdeveloped countries, which substantially delays rehabilitation procedures, interfering with the overall prognosis. One of the factors that contribute to this is the absence of a biomarker for the disease and the wide variety of diseases that can simulate and should be considered in the differential diagnosis., including later-onset, slower-progressing genetic diseases and neurometabolic genetic diseases, which may have a free-from-worsening interval. Demyelinating diseases can also have an atypical manifestation in childhood and interfere with this definition [5].
Another factor that may complicate the diagnosis of cerebral palsy in young children is that, in many cases, the clinical history does not show relevant antecedents or risk factors, such as pregnancy abnormalities, perinatal hypoxia, jaundice or prematurity. Especially in these patients, it would be important to carry out a more extensive approach or investigation, including genetic and metabolic screening [6].
NBIA is a rare disease, which most often begins in childhood. Prevalence data are not precise, with an estimated prevalence of between 1:500,000 and 1:1,000,000 inhabitants worldwide. Family history of multiple sclerosis may be associated with the occurrence of NBIA, as the disease commonly presents an increase in brain iron; however, the cause of this increase is still unclear [7]. It is known that there are 15 clinical-genetic forms and the most common are neurodegeneration associated with pantothenate kinase (PKAN), about 30–50% of cases, and phospholipase A2-associated neurodegeneration (PLAN), caused by mutations in the PLA2G6 gene [2, 8].
As demonstrated in a genetic test, the patient has the latter form, resulting from mutations in the PLA2G6 gene. This gene encodes a group of phospholipase A2 proteins, which are related to cell membrane homeostasis and also involved in the synthesis of fatty acids and lysophospholipids [9].
Regarding phenotypes, the PLAN form encompasses three overlapping types: (1) classic infantile neuroaxonal dystrophy (INAD); (2) atypical neuroaxonal dystrophy (AND); (3) late-onset dystonia-parkinsonism (PARK14) [10].
Onset of the INAD form occurs in childhood and manifests with progressive psychomotor deterioration, axial dystonia, spasticity, ataxia, and optic atrophy in some children. The progression of the disease is usually aggressive, causing death in the first decade of life [9]. However, the case described behaved differently in the clinical aspect (suggesting AND) having a slower progression and been confused with a non-progressive chronic encephalopathy.
This can be explained by the fact that there are non-progressive chronic encephalopathies that can manifest after a free interval and progress during a certain period, simulating a progressive encephalopathy. The opposite also occurs, when some metabolic or degenerative diseases can advance so slowly that they allow a certain degree of development, causing the impression of being a non-progressive encephalopathy [11].
Magnetic resonance imaging can direct to NBIA. In PLAN-type NBIA, there is usually disproportionate cortical and cerebellar atrophy. Iron deposition in the globus pallidus and substantia nigra may be less intense or absent in up to half of patients with the PKAN type. This was the case of our patient, who did not see the classic “tiger’s eye sign” (hypersignal area with hypointense halo in the globus pallidus) [12, 13].
In the present case, the diagnosis of Cerebral Palsy (CP) was initially given, a non-progressive neuromotor disorder that affects the development of movement, tone and posture due to a lesion in the developing brain that can occur from the fetal to post-natal period [14, 15]. In the patient’s clinical history, there was a case of hospitalization by neonatal fever, which was a confounding risk factor.
Cases of NBIA with a slowly progressive course may be more likely to be misdiagnosed as CP. It is important to pay attention to the main differential diagnoses that can delay the correct identification of a progressive neurodegenerative syndrome, causing a delay in adequate treatment. The family history and knowledge about the disease’s clinical evolution are important to find a correct differential diagnosis [11, 16].
There is still no recognized treatment for NBIA and gene therapy is still experimental. Some studies suggest that the use of the iron chelator deferiprone can slow down the PKAN form, with neuroimaging follow-up showing reduction in iron deposits. In the PLAN form there is little evidence that the antidepressant desipramine and deuterated polyunsaturated fatty acids can bring any benefit [8].
Conclusion
This report discusses the challenge of diagnosing neurological disorders that overlap in their clinical presentations. We emphasize the importance of a specific nosological diagnosis to direct genetic counseling, allowing the family to make the most assertive decision regarding the recurrence risk.
Availability of data and materials
Access to the data is restricted due to confidentiality between doctor and patient. Access to it must be consulted with the authors.
Abbreviations
- NBIA:
-
Brain iron accumulation neurodegeneration
- PLAN:
-
NBIA variant phospholipase A2
- PKAN:
-
Neurodegeneration associated with pantothenate kinase
- CP:
-
Cerebral Palsy
References
Nassif D, Pereira JS, Spitz M, Capitão C, Faria A. Neurodegeneration with brain iron accumulation: a case report. Dement Neuropsychol. 2016;10:160–4.
Salomão RPA, Pedroso JL, Gama MTD, Dutra LA, Maciel RH, Godeiro-Junior C, et al. A diagnostic approach for neurodegeneration with brain iron accumulation: clinical features, genetics and brain imaging. Arq Neuro-Psiquiatr. 2016;74:587–96.
Lehéricy S, Roze E, Goizet C, Mochel F. MRI of neurodegeneration with brain iron accumulation. Curr Opin Neurol. 2020;33:462–73.
Hayflick SJ, Kurian MA, Hogarth P. Neurodegeneration with brain iron accumulation. In: Handbook of clinical neurology. Amsterdam: Elsevier; 2018. p. 293–305.
Te Velde A, Morgan C, Novak I, Tantsis E, Badawi N. Early diagnosis and classification of cerebral palsy: an historical perspective and barriers to an early diagnosis. JCM 2019;8:1599.
Morgan C, Fahey M, Roy B, Novak I. Diagnosing cerebral palsy in full-term infants: cerebral palsy in infants. J Paediatr Child Health 2018;54:1159–64.
Schweser F, Hagemeier J, Dwyer MG, Bergsland N, Hametner S, Weinstock-Guttman B, et al. Decreasing brain iron in multiple sclerosis: the difference between concentration and content in iron MRI. Hum Brain Mapp. 2021;42:1463–74.
Iankova V, Karin I, Klopstock T, Schneider SA. Emerging disease-modifying therapies in neurodegeneration with brain iron accumulation (NBIA) disorders. Front Neurol. 2021;12:1–12.
Guo YP, Tang BS, Guo JF. PLA2G6-associated neurodegeneration (PLAN): review of clinical phenotypes and genotypes. Front Neurol. 2018;9:1–9.
Darling A, Aguilera-Albesa S, Tello CA, Serrano M, Tomás M, Camino-León R, et al. PLA2G6-associated neurodegeneration: new insights into brain abnormalities and disease progression. Parkinsonism Relat Disord. 2019;61:179–86.
Appleton RE, Gupta R. Cerebral palsy: not always what it seems. Arch Dis Child. 2019;104:809–14.
Lee JH, Yun JY, Gregory A, Hogarth P, Hayflick SJ. Brain MRI pattern recognition in neurodegeneration with brain iron accumulation. Front Neurol. 2020;11:1–9.
McNeill A, Birchall D, Hayflick SJ, Gregory A, Schenk JF, Zimmerman EA, et al. T2* and FSE MRI distinguishes four subtypes of neurodegeneration with brain iron accumulation. Neurology. 2008;70:1614–9.
Sadowska M, Sarecka-Hujar B, Kopyta I. Cerebral palsy: current opinions on definition, epidemiology, risk factors, classification and treatment options. NDT. 2020;16:1505–18.
Patel DR, Neelakantan M, Pandher K, Merrick J. Cerebral palsy in children: a clinical overview. Transl Pediatr. 2020;9:125–35.
Sewell MD, Eastwood DM, Wimalasundera N. Managing common symptoms of cerebral palsy in children. BMJ. 2014;349:1–13.
Acknowledgements
None.
Funding
This case report had no funding.
Author information
Authors and Affiliations
Contributions
All authors participated in all the steps regarding the confection of this article. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The research follows the ethical precepts described in law 466/12 and was approved by the Ethics and Research Committee under opinion number: 4.293.330/CAAE: 37212120.0.0000.8767. Consent from the patient and legal guardian was obtained as well.
Consent for publication
Consent from the patient and legal guardian was obtained.
Competing interests
The authors declares that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ferreira de Andrade, A., dos Santos Guimarães, G.K., Ferreira da Silva, J. et al. Neurodegeneration with brain iron accumulation: a differential diagnosis of cerebral palsy. Egypt J Neurol Psychiatry Neurosurg 59, 42 (2023). https://doi.org/10.1186/s41983-023-00639-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41983-023-00639-1