
Abstract
Background
Chordomas stem from notochordal vestiges and rank as low-grade bone malignancies although fraught with high risk of recurrence. This study assesses the clinical outcomes of twelve chordoma cases treated in our clinic, in an effort to shed light on the often under-represented pool of results deriving from non-referral centers.
Methods
We reviewed the clinicopathological traits of all chordoma patients registered in our center since 1991. Major endpoints were overall survival (OS) and progression-free survival (PFS) estimated using the Kaplan–Meier and Nelson–Aalen methods.
Results
Twelve patients, aged on average 47.9 years, were treated for primary or recurrent disease. Seven had chordomas originating in the cranium, 5 in the spine, including a bifocal tumor, and the mean time lapse between the beginning of symptoms and diagnosis was 15.4 months, marked by dull ache. Subtotal resection was achieved in 5 cases, incomplete in 5, while in 2, only biopsy was accomplished. Conformal radiotherapy was administered to 5 and stereotactic radiosurgery to 2 in the setting of recurrence. Protons were used once and targeted agents induced no clinical response in 3 patients. Median OS and PFS were 36 and 12 months, respectively, with the best outlook linked to maximal resection, spinal location, and good preoperative functional status. In all, 6 patients died of chordoma, 4 are alive, and 1 was lost. Relapse was the rule for most cases, except 2, and pulmonary metastases were ascertained in 1.
Conclusions
Our cases were typical of chordomas, implying that inadequate surgical margins and successive recurrence are negative determinants of prognosis and that interinstitutional cooperation counterbalances shortages in non-referral institutes.
Similar content being viewed by others

Introduction
Chordomas are tumors derived from the primitive notochord, a conformation of embryonic non-specialized cells that later develop mucin vacuoles, extending from the caudal end of the fetus to the future dorsum sellae and lying ventral to the neural tube [1]. The notochordal cells produce signaling proteins which, apart from driving cell differentiation of the neighboring tissues destined to constitute the neuro-spinal axis, are also thought to influence the patterning of the pancreas. During the fifth week of embryonic life, the degeneration of notochordal cells begins, resulting in them being embedded in the intervertebral space and representing the precursors of the “nucleus pulpocytes” found within the nucleus pulposus in adults [2]. Nevertheless, ectopic notochord remnants can be spread anywhere along the chord’s route, giving rise to a spectrum of conditions [3].
Tumors of the aforementioned cellular derivation exhibit either benign or malignant clinical behavior [4, 5]. Nowadays, according to the 4th edition of the WHO classification of soft tissue and bone tumors, notochordal neoplasms comprise of the intraosseous benign notochordal cell tumor and chordoma variants, with the latter being further categorized into classic chordoma, including the chordoid chordoma, dedifferentiated chordoma, and poorly differentiated chordoma [6]. According to today’s topography codes, chordomas are characterized as cranial, vertebral, and sacrococcygeal [7, 8]. Exceptionally, aberrant chordomas have been reported, inter alia, in the jaw, larynx, oropharynx, scapula, and knee [1, 9,10,11,12]. In contrast to the most prominent old series [1, 9, 13,14,15,16] indicating chordoma’s predilection for the caudal extremity of the axis, data from the US population-based Surveillance, Epidemiology, and End Results (SEER) program of the National Cancer Institute spanning the period 1973–2013, gave valuable clues about its actual spatial allocation, i.e., skull base (41.2%), true vertebrae (27.2%), sacrum (29.6%) [8].
Aside from myeloma, chordoma is the most common primary malignant bone tumor of the spine, accounting for 1–4% of all bone malignancies [6]. SEER data indicates an average annual incidence rate of 0.8 per million [17], and RARECARE project’s records indicate a crude incidence of 1 per million, with 218 new cases of chordoma arising per year [18]. Cranial chordomas’ 5- and 10-year survival rates have been reported to be 80.7% and 63.4%, respectively [19]. These results are indicative of survival increase in comparison with previous decades and consistent with the improved 10-year survival rate of 60% concerning extracranial chordomas [20]. An even more up-to-date study that embodied a subgroup of 39 skull base chordomas, reported a 93% 5-year survival attributed to advancements in diagnostic and treatment modalities [21].
Overall mean age at presentation for chordomas is 54.1 years [6], with younger patients faring better than older ones [17]. Despite chordomas’ low progression pace, patients finally succumb to multiple locoregional relapses, or to metastatic dissemination of tumor cells, mainly to the lungs, bones, lymph nodes, and liver in approximately 30–40% of all patients, predominantly in those terminally ill [22].
As an orphan disease, chordoma cases are dispersed across countries, hampering the ability to acquire large sample sizes and leading to underpowered statistics. A growing body of evidence stems from exemplar hospitals, equipped with modern technology and extensive interdisciplinarity infrastructure, with smaller centers being increasingly underrepresented, creating biased estimates for chordoma’s clinical progress. To compensate for that dearth of evidence, we report on our institution’s experience as a non-referral center.
Methods
Study design
To perform this study, we retrospectively considered all consecutive patients with histopathologically verified diagnosis of chordoma treated at the neurosurgery department of G. Papanikolaou General Hospital of Thessaloniki since 1991. All but one were followed until death or until June 2018. Therefore, this lost patient was excluded from survivorship analysis but, for full disclosure, his clinicopathological records and treatment applied are presented with the rest of the cohort.
We obtained approval from our center’s ethics board so that all relative medical charts, office documents, images, operative, and pathology reports could be retrieved. All procedures were in compliance with the 1964 Helsinki declaration and its later amendments. We extracted information on age, sex, symptoms, tumor location, and size as well as surgical margin evaluation. Direct examination or phone interviews using a standardized questionnaire were also conducted for additional follow-up data and were focused mainly on history of other hospitalizations, treatments, related complications, and the patients’ current functional status described via Karnofsky Performance Scale (KPS) and juxtaposed to the preoperative one.
Diagnosis and treatment strategy
We pursued single-stage surgeries or multi-staged ones in the case of large lesions infiltrating many anatomical compartments, provided that the patient’s desires, the comorbid medical conditions, and the accessibility of the tumor site did not deter it. Those patients were grouped into near total/subtotal (> 90% tumor) or incomplete/partial resection (> 50% tumor). Otherwise, extended biopsy (< 50% tumor) accompanied by palliative methods was the treatment of choice. Full consent was obtained by the patient and/or their family after ensuring that they were fully abreast of whatever the therapeutic procedures might have entailed. Diagnosis was substantiated in the majority of cases with immunohistochemistry techniques by the hospital’s neuropathologists. Microscopic slides of one uncertain case were sent for reassessment to the division of anatomic pathology of Aristotle University of Thessaloniki, which validated the diagnosis of chordoma.
Statistics
To assess survival outcome, we estimated the overall survival (OS) and the progression free-survival (PFS) rates. OS was defined as the time mediating between diagnosis and death or last follow-up, while PFS interval was calculated from the initiation of primary treatment to the time of radiologically visible expansion of known residuum, de novo relapse, or metastasis. When sequel images were not available, PFS was set as the period of symptomatic relief. OS and PFS were estimated using Kaplan–Meier survival curves to non-parametrically fit the survival function. We additionally used the Weibull model, by computing the rho and lambda parameters from our data and, since this model does not allow for non-positive durations, we converted one null value in the PFS duration in our samples to a miniscule one (1e-3). The Nelson–Aalen estimator was utilized to calculate the OS and PFS cumulative hazard rates. Cox’s Proportional Hazard model was deployed to explore the impact of the covariates on the survival rates and measure the regression coefficients. Pairwise differences between cohorts were gauged using the log rank test, identifying statistically significant associations of the examined variables. All statistical analyses were performed in python using the lifelines and pandas statistical packages and plotted using the matplotlib graphical package.
Results
Epidemiology
In the given time period, 12 patients affected by chordoma were enrolled, amounting to 13.6% of the total primary osseous tumors admitted to our ward in the same period. There were 10 men and 2 women ranging from 22 to 65 years of age (mean 47.9) at the time of diagnosis and all but one had primary chordomas. Most frequently affected sites were the cranium (n = 7), followed by the thoracic (n = 3), cervical (n = 2) and lumbar (n = 1) spine, with one of the cases presenting with a double localization in the T-11 and L-4 vertebral segments (Table 1).
Manifestation
Presenting symptoms were vague, unless the lesion was far advanced, and their mean duration until first consultation was 15.4 months, which reflects the typical indolent fashion of the disease’s clinical course. Specifically, pain was the preponderant symptom in every case, four patients also complained about visual disturbances (diplopia, field deficits, blurriness) with coincident exophthalmos in two, while one experienced rhinorrhea. Moreover, two patients sought medical advice on radiculopathy, one also encountered dysphagia, one felt a palpable neck mass, and hemiparesis appeared to another (Table 2). The mean KPS before surgery was 70 and was a function of the location and the extent of the tumor.
Pathology
Grossly, the resection specimens were described as friable, glistering, pinkish-gray masses often showing lobular clusters surrounded by a pseudocapsule, at low magnification. Microscopically, most of the cells making up the neoplasm were organized in cords, nests, and sheets within abundant myxoid matrix. The interpretations of histological sections were quite uniform and representative of classic chordoma, if one excludes a single identified “chondroid” variant, whose matrix bore resemblance to that of hyaline cartilaginous neoplasms. The characteristic physaliphorous cells with clear or mucin-filled intracytoplasmic droplets prevailed in most instances (10/12) over the smaller epithelioid cells and the sporadically seen spindled type, with a low mitotic activity, while, on occasion, areas of hemorrhage and necrosis were demonstrated.
The immunohistochemical detection of cytokeratins, epithelial membrane antigen (EMA), vimentin, and S100 protein in different levels on our tissue slides strengthens the notochordal hypothesis, yet staining for the recently recognized marker Brachyury was not attempted. The latter protein is encoded by the T gene, a member of the T-box gene family, activated in notochordal cells to regulate the transcription of genetic loci implicated in the developmental processes of the mesoderm, and pinpointed as overexpressed in chordomas [23].
Radiologic findings
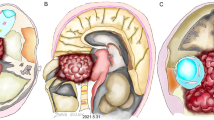
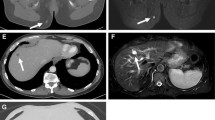
Roentgenograms and CT images were performed for all cases in our series with the former having miniscule diagnostic values in 6 cases. On plain radiographs, early-phase lesions could not be discerned, for all that, an expansile lytic bone lesion with a soft component that occupied contiguous anatomic areas was evident on CT scans. The radiolucent tumor permeated the cancellous bone but, of note, the adjacent soft tissue mass was disproportionally larger than the osseous deformity connoted. The MRI ushered in a new era in diagnostics and was available for 8 of our patients. As portrayed in the attached MRI slides (Figs. 1 and 2), chordomas are typified by intralesional septation mimicking a honeycomb, scattered calcifications, and heterogeneous contrast enhancement. On T1- and T2-weighted images, respectively, they show low and high signal intensities, and they are also hyperintense on diffusion sequence.

Case 7. A 60-year-old woman with primary cervical chordoma. Axial (a) and sagittal (b) MRI scans reveal C-4 vertebral body destruction associated with a large soft tissue mass. c Cervical spine X-ray after anterior C-4 corpectomy and reconstruction (titanium mesh cage, locking plate and screws)

Case 1. Preoperative (a) and postoperative (b) contrast MRI images. Last MRI scan (c) shows no disease progression
Treatment modes
Despite complete extirpation was never executed, on account of tumor transgression into neural structures, piecemeal near total resection was nonetheless the first treatment for five patients. Partial debulking surgery left gross disease behind in other five primarily treated tumors, while in two clival chordomas, extended endoscopic transsphenoidal biopsy was implemented. In the above initial procedures, intraoperative spillage could have occurred due to intralesional excision, potentially driving the 82% (n = 9) recurrence rate extrapolated in our cohort. Of those 9 that recurred, the two biopsied patients with one else were medically unfit for further surgery, and the remaining underwent 11 subsequent reoperations. In total, 4 combined two-staged approaches (n = 8), 1 combined three-staged approach (n = 3), and 18 solitary ones plus one ventriculoperitoneal shunt were performed, thus summing up to 30 operations, two of which took place abroad (Table 3).
In five cases, treatment was supplemented with megavoltage fractionated photon beam radiation and in two with stereotactic radiosurgery as a last resort and a way of ameliorating symptoms. Concomitant to radiotherapy, three advanced patients were pushed to the third treatment option of targeted agents such as imatinib and sirolimus, which did not bring off a slowdown in tumor proliferation. In our latest case, we proceeded to subtotal excision, augmented by proton radiotherapy to postpone recurrence, a goal that has been fulfilled since last year. We wanted to apply protons on two more chordomas, but implants hindered homogenous target dose distribution, and photon radiotherapy was instituted thereafter. In total, 5 combined staged approaches and 18 solitary ones plus one ventriculoperitoneal shunt were performed, thus summing up to 30 operations, two of which took place abroad (Table 3).
Complications
Eight adverse events complicated the course of five hospitalized patients in their lengthy recovery period, with the most serious ones being a cardiac arrest and a patient’s demise. Cases of postoperative neurologic impairment were minimized to only one owing to the surgeon’s judicious decision to respect the entrapped nerves at the expense of leaving tumor behind. Cerebrospinal fluid drainage obviated the need for wound revision in one case, though, another infected surgical wound unresponsive to conservative measures was perforce debrided. Regarding the remaining sequelae, the placement of a temporary tracheostomy was essential to tackle a clival lesion by the peroral approach, diabetes insipidus resolved automatically in one case and a last patient had a postoperative stroke (Table 4).
Survival outcome
The median length of life for this collective since the corroboration of the diagnosis was 36 months, while the median time to disease progression after a quiescent period by clinical and radiologic criteria was 12 months, standing for OS and PFS, respectively (Fig. 3). At the time of writing, seven people have died of massive disease, two are alive, still severely disabled, and two live with stable disease. All, barring the last two, eventually endured tumor recurrence, while one also presented with metastatic deposition to the lungs and thence they ultimately pursued supportive care. Of the survivors who maintain their independent status, one received protons and for the other, we elected to withhold radiation for use, presupposing that local relapse occurs. The completeness of excision, and high preoperative KPS independently predicted for better OS and PFS, whereas spinal chordomas afforded greater chance of PFS when collated with their intercranial counterpart (Fig. 4).

Life table analysis determined OS and PFS using the Kaplan–Meier (a, c) and Nelson–Aalen estimators (b, d)

Survival curves in relation to risk factors (a) OS by resection, b PFS by resection, c OS by site, d PFS by site, e OS by pre-op KFS, and f PFS by pre-op KPS
Discussion
In this study, we document a series of 12 chordoma cases through a single institution’s experience in Greece. Undoubtedly the common practice in many medical centers deviates from the ideal due to deficits in resources, use of outdated technology, or lack of surgical experience. The major objective of the analysis was to provide insight into the treatment of the disease and a framework for better practice in the future after an overview of the clinical and paraclinical characteristics of our patients and their therapeutic outcomes.
Due to the inherent biology of the tumor, chordomas’ clinical management has been traditionally dependent upon operative control. Boriani claimed aptly that “treatment of spinal chordoma is a 1-shot surgery” to accent the importance of the first surgery and the futility of reoperations in the long term [24]. Clarke reached a similar conclusion emphasizing the beneficial effect of maximal excisions of tumors not previously violated and proposed to include even the biopsy needle tract in the surgery plan to avoid recurrence [25]. Enneking in his surgical staging system for musculoskeletal sarcomas described operative margins as radical, wide, marginal, or intralesional [26]. However, due to the relentless progression of chordomas and their proximity to critical structures, gross total resection with free “wide” margins seems elusive even in the case of en bloc sacrectomy procedures [25], and this is the reason we only refer to intralesional excisions in this study. Particularly, when it comes to cervical chordomas, unless they are restricted within the vertebral body, striving for ample margins can irreversibly harm the patient [27].
Balancing between operative radicality and postoperative functional neurological status under the threat of recurrence and the inevitable fatal termination is challenging. Equally difficult is the decision to cease re-operating, an issue born out in our series and others [28] that necessitate good judgement on the surgeon’s side. The 82% rate of recurrence cited herein is daunting and credited largely to incomplete surgeries not outweighed by modern radiotherapy. In hindsight, it could have been more beneficial for our patients to carry out more radical operations, decrease the surgical stages, and select more suitable approaches in the context of a personalized treatment plan.
The dura, until breached, impedes early tumor expansion; hence, the worth of an epidural route has emerged from the literature [29, 30]. Sen failed to evince superiority among an assortment of either lateral or midline anterior or combined approaches to the skull base in a total of 80 operations but accentuated the positives of the recent wide application of endoscopic endonasal approach (EEA) [30], which seems to gain momentum according to many studies [31,32,33,34]. In a series of 54 patients harboring skull base chordomas managed exclusively with EEA, Chibbaro recorded a gross-total resection rate of 65% and underlined the method’s shortcoming in repairing the base’s defect [32]. Gui, having experience with 161 cases, separated cranial chordomas into anterior skull base type, superior clivus type, superior-middle clivus type, middle-inferior clivus type, inferior clivus type, total clivus type, and extensive type (tumor in the median and paramedian regions) to direct the surgical corridor through the EEA and suggested a subsequent phase II open approach or just even a single craniotomy to facilitate resection of those too far laterally, superiorly, or inferiorly located tumors [33].
The advent of particle therapy paired with the evolution of molecular-targeted drugs and the tremendous advancements in terms of surgical approaches, reconstruction, and microsurgical techniques have reinvigorated the inception of a potent cure against an infiltrative radioresistant tumor that poorly responds to cytotoxic chemotherapy. Refinements in the field of neuroimaging, neuroanesthesia, neurointensive medicine and the widespread adoption of intraoperative neuronavigation and electrophysiological monitoring also played a contributory role. This paradigm of multimodal treatment, in the concept of maximally safe cytoreductive surgery, has many proponents [35, 36], but still in this series due to the small number of patients, selection bias, and diversity of radiation modalities; no definitive conclusions on the benefits from adjuvant treatment regimens could be drawn.
The adjuvant treatment mostly advocated in the literature, albeit with the pertinent discrepancies, is proton beam radiotherapy, which is capable of delivering high-energy particles to the tumor bed with precision, ergo lessening the dose to tissues in close vicinity [22, 37]. Rotondo studied the preoperative administration of proton-based radiotherapy (RT) to 44 primary and 16 recurrent spinal chordomas followed by a boost postoperative RT and deduced 5-year overall survival (OS) rates of 85% for the primary chordomas and 71% for the recurrent ones [38]. Likewise, 91 skull base chordomas attained good outcomes when given mixed proton–photon irradiation with the prescribed dose adjusted to postoperative tumor volume, a value of which less than 25 ml conferred higher local control rates (LC) [39]. Imai surveyed the efficacy of carbon ion radiotherapy for 95 unresectable sacral chordomas and observed a 5-year overall survival rate at 86% [40], which is in keeping with the favorable results shown by Uhl in 56 similar cases irradiated either solely with carbon ions or in combination with photon IMRT [41]. Uhl et al. also accumulated the results of 155 skull base chordomas treated with carbon ions, yielding at 5 years an OS rate of 85% and a corresponding LC rate of 72% [42]. Nowadays, a phase III trial is ongoing by Uhl’s associates with the purpose of sorting out whether the higher relative biological effectiveness of carbon ions reflects a therapeutic advantage over protons [43].
A short time ago, we came across the dilemma of modifying the fixation system of a patient, suffering from gross residual disease of the thoracic spine, since questions had been raised as to whether the target volume could be delineated and covered sufficiently in the presence of metal-induced artifacts, leading to him being rejected from two proton therapy centers. Unfortunately, a few months later, he developed signs of impending paraplegia, and a thoracotomy was performed, the third overall, with the intent of anterior spinal cord decompression, leaving the instrumentation intact and followed by stereotactic radiosurgery (SRS). Staab et al. in a prior work implied that surgical stabilization (SS) could possibly compromise the outcome of extracranial chordomas, quoting a 100% actuarial LC rate at 5 years for 19 patients without SS, after actively scanned proton delivery. On the opposite to this cohort, the same radiation technique heralded a clearly worse 5-year LC rate of 30% when used on another subgroup of 21 patients who required titanium-based SS [44].
Considering small intracranial tumors, SRS is an effective alternative to particles correlated with fewer adverse radiation effects (AREs), as evidenced in a study embodying 71 cranial chordomas, out of whom 51 that had not received fractionated radiation therapy prior to SRS trended toward better 5-year survival rates contrary to the rest (93% vs 43%) [45]. In another series with a median follow-up time of 34 months, tumor control was the fact for 8/16 patients with cranial chordomas who had an operation in conjunction with SRS [46]. SRS for spinal chordomas is not that well-established but has clinical merits warranting evaluation in view of the absence of particle therapy facilities in countries that render the treatment cost prohibitive [47, 48].
The refractoriness of chordoma cells to cytotoxic chemotherapy and their rich expression of numerous tyrosine kinase receptors, such as PDGFR-α, PDGFR-β, EGFR, VEGFR, HER2, KIT, and MET prompted the appraisal of molecular inhibitors as novel therapeutics in chordomas. So far, imatinib, sunitinib, sorafenib and lapatinib have been tested in phase II trials with appreciable results on disease remission [49], which is in striking contrast with the unpleasant inferences made about our three eligible medicated patients.
Published records to date report a mean age at diagnosis of 47.5 for chordomas arising in the basiocciput [19], and 59.9 years for the extracranial lesions [20]. In our cohort, the mean age for cranial lesions was 46.9 and for extracranial was 49.4. In particular, the youngest patients in our series were one 22- and one 24-year-old males afflicted with spheno-occipital chordoma, which is in line with the reported age spectrum of the disease in regard with location [13].
The etiologic factors of the dismal survival outcomes in our series could be attributed to delayed presentation to physician, contaminated margins, and iatrogenic tumor seeding, portending primary site failure. Palliative conventionally fractionated radiotherapy appeared to relieve pain and restrain tumor regrowth only temporarily, an experience shared by others before the emergence of proton therapy [10, 13, 50,51,52]. The longest survivor is successfully salvaged at 7 years and harbors a cervical chordoma with cartilaginous differentiation, which concurs with Heffelfinger’s perspective on that pathology [14]. Only two cases of distant failures were discovered, but others could have been missed, owing to staging or follow-up procedures that did not encompass all risky sites.
Methodologic limitations and recommendations
Emphasis should also be put on the limitations of this study, given the rarity of this disease that precludes not only an individual clinician’s potential to expertise but also a center’s ability to output large series and offer a rationale for treatment. For its retrospective nature, the present analysis could be susceptible to bias, emanating from restrictions related to patient heterogeneity, individually tailored protocols, changes in surgical team, and improvements in practices over the last 27 years. Further, we relied on scant data on some patients, especially from the earlier era without omitting those who had a short follow-up. This article depicts the outcomes of 12 patients subjected to a non-subspecialized center and, despite not reaching statistical significance, they were indeed illustrative of all clinic-anatomical features of chordoma and could be examined on their concordance with other series.
Due to its peculiarities, this condition is not amenable to satisfactory treatment and still follows a set fatal course. The position paper of the Chordoma Global Consensus Group in January 2015 concludes that in order to optimize treatment outcomes, chordoma patients should be served in specialized centers with a high caseload and a multitude of disciplines involved [22]. On the basis of the second consensus group meeting on how to tackle locoregional recurrence of chordoma, convened in November 2015, when metal implants cannot be altered, extracted, or replaced with carbon fiber devices, photons, which generate less artifacts, can be delivered in lieu of charged particle radiotherapy [18]. In countries deprived of referral institutes, surgical hospitals of average size such as ours, that prefer to follow a safe rather than an aggressive treatment policy, are in imperative need of interdisciplinary communication with radiotherapists and oncologists of other domestic or even foreign institutions in order to prevent instrumentation-caused interference to radiotherapy plans and enable patient participation in coordinated experimental research programs.
Conclusions
A review of 12 chordoma cases was undertaken, confirming the well-known poor prognosis. The cases described in our study hint that intralesional excision and the ensuing recurrence compromise the outcome. To remedy this, the study features the necessity for interdisciplinary communication in non-referral centers. Notably, the inability to compile a reasonable number of samples and, therefore, to make high-confidence statistical inferences is dictated by the rarity of the disease. It is the authors’ contention that a large prospective multicenter Greek study poses a great challenge and as such, this series is submitted to be added to the previously published ones and act altogether as historical controls for future studies.
Availability of data and materials
All data used for this analysis are included in the manuscript tables.
Abbreviations
- OS:
-
Overall survival
- PFS:
-
Progression-free survival
- STS:
-
Stereotactic radiosurgery
- KPS:
-
Karnofsky Performance Scale
- EEA:
-
Endoscopic endonasal approach
- AREs:
-
Adverse radiation effects
- PDGFR:
-
Platelet-derived growth factor receptor
- EGFR:
-
Epidermal growth factor receptor
- VEGFR:
-
Vascular endothelial growth factor receptor
- CCJ:
-
Craniocervical junction
- WBB:
-
Weinstein-Boriani-Biagini
- DOS:
-
Duration of symptoms
- m:
-
Months
- LC:
-
Local control
- NTR:
-
Near total resection
- NEP:
-
No evidence of progression
- APD:
-
Alive with progressive disease
- DM:
-
Distant metastasis
- DPD:
-
Dead of progressive disease
- OpD:
-
Operative death
- NR:
-
Not reported
- CT:
-
Computed tomography
- MRI:
-
Magnetic resonance imaging
References
Faust DB, Gilmore HR, Mudgett CS. Chordomata: a review of the literature, with report of a sacrococcygeal case. Ann Intern Med. 1944;21:678–98.
Ramesh T, Nagula SV, Tardieu GG, Saker E, Shoja M, Loukas M, et al. Update on the notochord including its embryology, molecular development, and pathology: a primer for the clinician. Cureus. 2017;9:e1137. https://doi.org/10.7759/cureus.1137.
Horwitz T. Chordal ectopia and its possible relation to chordoma. Arch Pathol. 1941;31:354–62.
Zulch K. Biologie und pathologie der hirngeschwulste. Handbuch Der Neurochirurgie. 1956;3:467.
Pamir M, Al-Mefty O, Borba L. Chordomas: technologies, techniques, and treatment strategies: Thieme; 2017.
Bocklage T, Quinn R, Schmit B, Verschraegen C. Bone and soft tissue tumors: a multidisciplinary review with case presentations: JP Medical Limited; 2014.
Coenen H. Das chordom. Bruns’ Beitr Klin Chir. 1925;133:1–77.
Lee IJ, Lee RJ, Fahim DK. Prognostic factors and survival outcome in patients with chordoma in the United States: a population-based analysis. World Neurosurg. 2017;104:346–55. https://doi.org/10.1016/j.wneu.2017.04.118.
Mabrey RE. Chordoma: a study of 150 cases. Am J Cancer. 1935;25:501–17.
Higinbotham NL, Phillips RF, Farr HW, Hustu HO. Chordoma. thirty-five-year study at memorial hospital. Cancer. 1967;20(11):1841–50. https://doi.org/10.1002/1097-0142(196711)20:11<1841::AID-CNCR2820201107>3.0.CO;2-2.
Li X, Wang Y, Wang F, Li B, Sun S, Yang H. An unusual case of oropharyngeal chordoma: a case report and literature review. Medicine. 2017;96(48):e8963. https://doi.org/10.1097/MD.0000000000008963.
Paweł K, Magdalena F, Michał D, Larque BA, Petur NG. Primary extra-axial, para-articular chordoma of the knee. A case report and the review of literature. Histopathology. 2018;72(5):883–5. https://doi.org/10.1111/his.13440.
Dahlin DC, Maccarty CS. Chordoma. A study of fifty-nine cases. Cancer. 1952;5(6):1170–8. https://doi.org/10.1002/1097-0142(195211)5:6<1170::AID-CNCR2820050613>3.0.CO;2-C.
Heffelfinger MJ, Dahlin DC, Maccarty CS, Beabout JW. Chordomas and cartilaginous tumors at the skull base. Cancer. 1973;32(2):410–20. https://doi.org/10.1002/1097-0142(197308)32:2<410::AID-CNCR2820320219>3.0.CO;2-S.
Eriksson B, Gunterberg B, Kindblom LG. Chordoma: a clinicopathologic and prognostic study of a Swedish national series. Acta Orthop. 1981;52(1):49–58. https://doi.org/10.3109/17453678108991758.
O’Neill P, Bell B, Miller J, Jacobson I, Guthrie W. Fifty years of experience with chordomas in southeast Scotland. Neurosurgery. 1985;16(2):166–70. https://doi.org/10.1227/00006123-198502000-00007.
Smoll NR, Gautschi OP, Radovanovic I, Schaller K, Weber DC. Incidence and relative survival of chordomas: the standardized mortality ratio and the impact of chordomas on a population. Cancer. 2013;119(11):2029–37. https://doi.org/10.1002/cncr.28032.
Stiller CA, Trama A, Serraino D, Rossi S, Navarro C, Chirlaque MD, et al. Descriptive epidemiology of sarcomas in Europe: report from the RARECARE project. Eur J Cancer. 2013;49(3):684–95. https://doi.org/10.1016/j.ejca.2012.09.011.
Chambers KJ, Lin DT, Meier J, Remenschneider A, Herr M, Gray ST. Incidence and survival patterns of cranial chordoma in the United States. Laryngoscope. 2014;124(5):1097–102. https://doi.org/10.1002/lary.24420.
Mukherjee D, Chaichana KL, Gokaslan ZL, Aaronson O, Cheng JS, McGirt MJ. Survival of patients with malignant primary osseous spinal neoplasms: results from the Surveillance, Epidemiology, and End Results (SEER) database from 1973 to 2003. J Neurosurg Spine. 2011;14(2):143–50. https://doi.org/10.3171/2010.10.SPINE10189.
Di Maio S, Rostomily R, Sekhar LN. Current surgical outcomes for cranial base chordomas: cohort study of 95 patients. Neurosurgery. 2012;70(6):1355–60. https://doi.org/10.1227/NEU.0b013e3182446783.
Stacchiotti S, Sommer J. Building a global consensus approach to chordoma: a position paper from the medical and patient community. Lancet Oncol. 2015;16(2):e71–83. https://doi.org/10.1016/S1470-2045(14)71190-8.
Zhu J, Kwan KM, Mackem S. Putative oncogene brachyury (t) is essential to specify cell fate but dispensable for notochord progenitor proliferation and eMT. Proc Natl Acad Sci. 2016;113(14):3820–5. https://doi.org/10.1073/pnas.1601252113.
Boriani S, Bandiera S, Biagini R, Bacchini P, Boriani L, Cappuccio M, et al. Chordoma of the mobile spine: fifty years of experience. Spine. 2006;31(4):493–503. https://doi.org/10.1097/01.brs.0000200038.30869.27.
Clarke MJ, Dasenbrock H, Bydon A, Sciubba DM, McGirt MJ, Hsieh PC, et al. Posterior-only approach for en bloc sacrectomy: clinical outcomes in 36 consecutive patients. Neurosurgery. 2012;71(2):357–64. https://doi.org/10.1227/NEU.0b013e31825d01d4.
Enneking WF, Spanier SS, Goodman MA. A system for the surgical staging of musculoskeletal sarcoma. Clin Orthop Relat Res. 1980;153:106–20.
Wang Y, Xiao J, Wu Z, Huang Q, Huang W, Zhu Q, et al. Primary chordomas of the cervical spine: a consecutive series of 14 surgically managed cases: clinical article. J Neurosurg Spine. 2012;17:292–9. https://doi.org/10.3171/2012.7.SPINE12175.
Choi D, Melcher R, Harms J, Crockard A. Outcome of 132 operations in 97 patients with chordomas of the craniocervical junction and upper cervical spine. Neurosurgery. 2010;66(1):59–65. https://doi.org/10.1227/01.NEU.0000362000.35742.3D.
Takahashi S, Kawase T, Yoshida K, Hasegawa A, Mizoe JE. Skull base chordomas: efficacy of surgery followed by carbon ion radiotherapy. Acta Neurochir. 2009;151(7):759–69. https://doi.org/10.1007/s00701-009-0383-5.
Sen C, Triana AI, Berglind N, Godbold J, Shrivastava RK. Clival chordomas: clinical management, results, and complications in 71 patients. J Neurosurg. 2010;113(5):1059–71. https://doi.org/10.3171/2009.9.JNS08596.
Koutourousiou M, Gardner PA, Tormenti MJ, Henry SL, Stefko ST, Kassam AB, et al. Endoscopic endonasal approach for resection of cranial base chordomas: outcomes and learning curve. Neurosurgery. 2012;71(3):614–24. https://doi.org/10.1227/NEU.0b013e31825ea3e0.
Chibbaro S, Cornelius JF, Froelich S, Tigan L, Kehrli P, Debry C, et al. Endoscopic endonasal approach in the management of skull base chordomas - clinical experience on a large series, technique, outcome, and pitfalls. Neurosurg Rev. 2014;37(2):217–24. https://doi.org/10.1007/s10143-013-0503-9.
Gui S, Zong X, Wang X, Li C, Zhao P, Cao L, et al. Classification and surgical approaches for transnasal endoscopic skull base chordoma resection: a 6-year experience with 161 cases. Neurosurg Rev. 2016;39(2):321–33. https://doi.org/10.1007/s10143-015-0696-1.
Rahme RJ, Arnaout OM, Sanusi OR, Kesavabhotla K, Chandler JP. Endoscopic approach to clival chordomas: the northwestern experience. World Neurosurg. 2018;110:e231–8. https://doi.org/10.1016/j.wneu.2017.10.146.
Walcott BP, Nahed BV, Mohyeldin A, Coumans JV, Kahle KT, Ferreira MJ. Chordoma: current concepts, management, and future directions. Lancet Oncol. 2012;13(2):e69–76. https://doi.org/10.1016/S1470-2045(11)70337-0.
Jägersberg M, El Rahal A, Dammann P, Merkler D, Weber DC, Schaller K. Clival chordoma: a single-centre outcome analysis. Acta Neurochir. 2017;159(10):1815–23. https://doi.org/10.1007/s00701-017-3163-7.
Yasuda M, Bresson D, Chibbaro S, Cornelius JF, Polivka M, Feuvret L, et al. Chordomas of the skull base and cervical spine: clinical outcomes associated with a multimodal surgical resection combined with proton-beam radiation in 40 patients. Neurosurg Rev. 2012;35(2):171–82. https://doi.org/10.1007/s10143-011-0334-5.
Rotondo RL, Folkert W, Liebsch NJ, Chen Y-LE, Pedlow FX, Schwab JH, et al. High-dose proton-based radiation therapy in the management of spine chordomas: outcomes and clinicopathological prognostic factors. J Neurosurg Spine. 2015;23(6):788–97. https://doi.org/10.3171/2015.3.SPINE14716.
Fung V, Calugaru V, Bolle S, Mammar H, Alapetite C, Maingon P, et al. Proton beam therapy for skull base chordomas in 106 patients: a dose adaptive radiation protocol. Radiother Oncol. 2018;128(2):198–202. https://doi.org/10.1016/j.radonc.2017.12.017.
Imai R, Kamada T, Sugahara S, Tsuji H, Tsujii H. Carbon ion radiotherapy for sacral chordoma. Br J Radiol. 2011;84(special_issue_1):48–53. https://doi.org/10.1259/bjr/13783281.
Uhl M, Welzel T, Jensen A, Ellerbrock M, Haberer T, Jäkel O, et al. Kohlenstoffionenbestrahlung von Patienten mit primären und rezidivierten sakralen Chordomen. Strahlenther Onkol. 2015;191(7):597–603. https://doi.org/10.1007/s00066-015-0825-3.
Uhl M, Mattke M, Welzel T, Roeder F, Oelmann J, Habl G, et al. Highly effective treatment of skull base chordoma with carbon ion irradiation using a raster scan technique in 155 patients: first long-term results. Cancer. 2014;120(21):3410–7. https://doi.org/10.1002/cncr.28877.
Nikoghosyan AV, Karapanagiotou-Schenkel I, Münter MW, Jensen AD, Combs SE, Debus J. Randomised trial of proton vs. carbon ion radiation therapy in patients with chordoma of the skull base, clinical phase III study HIT-1-Study. BMC Cancer. 2010;10:607. https://doi.org/10.1186/1471-2407-10-607.
Staab A, Rutz HP, Ares C, Timmermann B, Schneider R, Bolsi A, et al. Spot-scanning-based proton therapy for extracranial chordoma. Int J Radiat Oncol Biol Phys. 2011;81(4):489–96. https://doi.org/10.1016/j.ijrobp.2011.02.018.
Kano H, Iqbal FO, Sheehan J, Mathieu D, Seymour ZA, Niranjan A, et al. Stereotactic radiosurgery for chordoma: a report from the North American Gamma knife consortium. Neurosurgery. 2011;68(2):379–88. https://doi.org/10.1227/NEU.0b013e3181ffa12c.
Förander P, Bartek J, Fagerlund M, Benmaklouf H, Dodoo E, Shamikh A, et al. Multidisciplinary management of clival chordomas; long-term clinical outcome in a single-institution consecutive series. Acta Neurochir. 2017;159(10):1857–68. https://doi.org/10.1007/s00701-017-3266-1.
Yamada Y, Laufer I, Cox BW, Lovelock DM, Maki RG, Zatcky JM, et al. Preliminary results of high-dose single-fraction radiotherapy for the management of chordomas of the spine and sacrum. Neurosurgery. 2013;73(4):673–80. https://doi.org/10.1227/NEU.0000000000000083.
Jung EW, Jung DL, Balagamwala EH, Angelov L, Suh JH, Djemil T, et al. Single-fraction spine stereotactic body radiation therapy for the treatment of chordoma. Technol Cancer Res Treatment. 2017;16(3):302–9. https://doi.org/10.1177/1533034616652775.
Colia V, Stacchiotti S. Medical treatment of advanced chordomas. Eur J Cancer. 2017;83:220–8. https://doi.org/10.1016/j.ejca.2017.06.038.
Fuller DB, Bloom JG. Radiotherapy for chordoma. Int J Radiat Oncol Biol Phys. 1988;15(2):331–9. https://doi.org/10.1016/S0360-3016(98)90012-8.
Romero J, Cardenes H, la Torre A, Valcarcel F, Magallon R, Regueiro C, et al. Chordoma: results of radiation therapy in eighteen patients. Radiother Oncol. 1993;29(1):27–32. https://doi.org/10.1016/0167-8140(93)90169-9.
Zorlu F, Gürkaynak M, Yildiz F, Öge K, Atahan IL. Conventional external radiotherapy in the management of clivus chordomas with overt residual disease. Neurol Sci. 2000;21(4):203–7. https://doi.org/10.1007/s100720070077.
Acknowledgements
Not applicable
Funding
No funding was received for this research.
Author information
Authors and Affiliations
Contributions
MK coordinated and planned the analysis and authored the article. FT carried out the statistical analysis of the data and contributed to the manuscript. PS was the head responsible for the surgical management for the majority of the patients and contributed to the manuscript. IB was the principal investigator and supervisor of the study and contributed to the manuscript. All authors have read and approve submission of this manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the ethics committee of the George Papanikolaou General Hospital of Thessaloniki with approval number 4/3.1.2018. The patients provided verbal consent. The committee’s reasoning was that since this is a retrospective study into the hospital’s archives, formal consent is not required. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Consent for publication
Not applicable
Competing interests
All authors certify that they have no affiliations with or involvement in any organization or entity with any financial interest (such as honoraria; educational grants; participation in speakers’ bureaus; membership, employment, consultancies, stock ownership, or other equity interest; and expert testimony or patent-licensing arrangements), or non-financial interest (such as personal or professional relationships, affiliations, knowledge, or beliefs) in the subject matter or materials discussed in this manuscript.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Karampouga, M., Tsetsos, F., Sakellariou, P. et al. Outcomes and issues of 12 chordomas treated in a single center. Egypt J Neurol Psychiatry Neurosurg 57, 52 (2021). https://doi.org/10.1186/s41983-021-00306-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41983-021-00306-3