
Abstract
Background
Cutaneous melanoma (CM) is the most aggressive subtype of skin cancer, with increasing incidence over the past several decades. DNA methylation is a key element of several biological processes such as genomic imprinting, cell differentiation and senescence, and deregulation of this mechanism has been implicated in several diseases, including cancer. In order to understand the relationship of DNA methylation in CMs, we searched for an epigenetic signature of cutaneous melanomas by comparing the DNA methylation profiles between tumours and benign melanocytes, the precursor cells of CM.
Methods
We used 20 primary CMs and three primary cell cultures of melanocytes as a discovery cohort. The tumours mutational background was collected as previously reported. Methylomes were obtained using the HM450K DNA methylation assay, and differential methylation analysis was performed. DNA methylation data of CMs from TCGA were recovered to validate our findings.
Results
A signature of 514 differentially methylated genes (DMGs) was evident in CMs compared to melanocytes, which was independent of the presence of driver mutations. Pathway analysis of this CM signature revealed an enrichment of proteins involved in the binding of DNA regulatory regions (hypermethylated sites), and related to transmembrane signal transducer activities (hypomethylated sites). The methylation signature was validated in an independent dataset of primary CMs, as well as in lymph node and distant metastases (correlation of DNA methylation level: r > 0,95; Pearson’s test: p < 2.2e-16).
Conclusions
CMs exhibited a DMGs signature, which was independent of the mutational background and possibly established prior to genetic alterations. This signature provides important insights into how epigenetic deregulation contributes to melanomagenesis in general.
Similar content being viewed by others

Background
DNA methylation is the covalent addition of methyl radicals at nucleotides, mainly at CpG sites, which is catalysed by the DNA methyltransferase (DNMT) enzyme family. In the context of gene promoters enriched in CpG dinucleotides, this epigenetic mark is consistently associated with the stable repression of gene expression [1]. DNA methylation is a key element of several biological processes such as genomic imprinting, cell differentiation and senescence, and deregulation of this mechanism has been implicated in several diseases, including cancer [1].
Cutaneous melanoma (CM) is the most aggressive subtype of skin cancer, with increasing incidence over the past several decades [2]. Efforts to understand the molecular biology of CM has led to improvements in the clinic, such as the discovery of recurrent mutations in the MAP kinase pathway, which are responsible for the transduction of proliferative extracellular signals. A hotspot mutation in the BRAF oncogene (BRAFV600E) is a target for selective inhibitors, which has been shown to improve the overall and progression-free survival of a group of patients [3].
In addition to screening for driver mutations in CMs for treatment management, data on epigenetic deregulation have also been translated to the clinic: the gain of methylation in a panel of genes is associated with increasing tumour stage [4], and the presence of methylated genes in the serum of CM patients can predict responses to biochemotherapy and disease outcome [5].
Recently, Hou et al. observed that melanoma cell lines harbouring BRAFV600E mutations undergo major DNA methylation alterations (both hypomethylation and hypermethylation events) upon BRAF knockdown [6]. In a previous attempt to integrate the genetic and epigenetic alterations, our group also revealed an association between hypomethylation of the repetitive element LINE-1 and the presence of CDKN2A inactivation in primary CMs [7].
The aim of the present work was to investigate whether CMs exhibit an epigenetic signature, i.e., a set of epigenetic markers, independent of the presence of specific genetic mutations.
Methods
Tumour samples and DNA extraction
Twenty primary CM samples and seven metastatic tissues (all of them fresh frozen tissues) were retrieved from the AC Camargo Cancer Center Biobank (ACCCC, Sao Paulo, Brazil). Samples contained >80% of tumour cells and an absence of necrotic regions and/or inflammatory infiltrate. This project was approved by the ethics committee of the institution (CEP ACCCC 1765/13). Clinical characterization of tumour samples is provided in Additional file 1: Figure S1. The presence of melanoma driver mutations were previously investigated in these CMs [7] and are described in Additional file 1: Figure S1.
Three primary cultures of melanocytes isolated from the foreskin of healthy donors [8, 9] were used as controls. The primary cultures were obtained in collaboration with the School of Pharmaceutical Sciences from the University of Sao Paulo (University of Sao Paulo – CEP HU/USP 943/09), and DNA samples were extracted from early passages.
Whole-genome DNA methylation analysis
Approximately 500 ng of bisulfite-converted DNA from each sample was used for analysis. We assessed the whole-genome DNA methylation level of all samples using an Infinium HumanMethylation450 BeadChip Kit (HM450K) (Illumina), according to the manufacturer’s protocol.
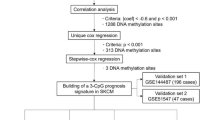
Data analysis was performed using an in-house pipeline running in R software. Briefly, a pre-processing step was performed, excluding probes with a detection p-value >0.01, those mapped at sex chromosomes, those containing Single Nucleotide Polymorphisms (SNPs), or those mapping to multiple sites. Normalization was then performed using the beta mixture quantile dilation method [10] with conversion to M values. Using the median of the methylation values of the CpG probes mapped at promoter regions (e.g., 1500 nucleotides from the transcription start site TSS1500; 200 nucleotides from the transcription start site TSS200; 5’UTR; and 1st exon), a single methylation value was consolidated for each gene promoter represented in the HM450K platform. We used the Wilcoxon signed rank test (two-sided) with a 1% false discovery rate correction to detect significant DNA methylation differences between groups. We then calculated the difference of median methylation levels between groups to obtain the differential methylation value (ΔM) for a given gene promoter between two given groups.
After each comparison between tumour groups harbouring different mutations and the control group of melanocytes, we intersected the differentially methylated sites detected in the different comparisons, thus generating a list of commonly differentially methylated genes (DMGs). The list of DMGs was analysed using WebGestalt software [11] with the following default analysis parameters: Gene Ontology (GO) enrichment and hypergeometric test with Benjamini and Hochberg correction to identify statistically significant enrichments (p < 0,01).
In silico analysis using publicly available data on DNA methylation from TCGA melanoma samples
We retrieved DNA methylation values of the genes determined to be differentially methylated in an independent cohort of primary cutaneous melanomas using HM450K level 3 data deposited in TCGA (https://cancergenome.nih.gov/). Methylation data from 91 primary CMs were retrieved, and also from 259 lymph node metastatic tissues and 48 tissues from distant metastases. We also used our in house pipeline to analyse the TCGA data set.
We then calculated the correlation of the methylation levels of these gene promoters between our CM samples and TCGA data using the sites of the detected signature of differentially methylated genes. We utilized the cor.test function of R statistical software (https://www.r-project.org/) and applied the Pearson’s test to evaluate the significance of the correlation. We also applied a bootstrap analysis, in which 500 genes were randomly sampled several times, and their methylation levels correlated with the methylation levels from the abovementioned datasets.
Results
A set of 514 differentially methylated genes constitute an epigenetic signature for primary cutaneous melanomas
As previously reported [7], this group of CMs is composed of 12 tumours harbouring CDKN2A inactivation; twelve tumours carried TERT promoter mutations, one sample had a KIT mutation, four samples had NRAS mutations, and seven had BRAF mutations. Three of the 20 tumours were wild-type for the investigated alterations.
A total of 1396 DMG promoters were detected in CMs compared to control melanocytes, the majority of which (1039) were hypermethylated. Differential methylation analysis against control melanocytes was then performed to group the CMs according to their mutational profile. In all cases, a substantial number of DMGs was obtained (Additional file 2: Table S1).
We intersected all DMGs presenting with more than 10% methylation differences between CMs and control melanocytes. A common set of 514 DMGs was obtained, which was independent of the mutational background of the primary CM (Additional file 3: Table S2). We then classified these 514 genes into either hypermethylated or hypomethylated for a GO enrichment analysis that was performed using WebGestalt online software [11]. Hypermethylated genes were enriched in functions involved in the binding of transcription factors to specific DNA sequences, whereas hypomethylated genes were related to transmembrane receptor and signal transducer activities (Additional file 4: Table S3).
Validation of the signature 514 DMGs using an independent cohort of CMs
To determine whether the detected epigenetic signature of 514 DMGs was a finding restricted to our group of tumours, we retrieved HM450K data from 91 primary CMs deposited in TCGA. A high correlation of the methylation levels of the 514 gene promoters was observed upon comparing our CM group to TCGA melanoma samples (r = 0.90), whereas the correlation of promoter DNA methylation level from TCGA data and control melanocytes was lower, as expected (r = 0.63) (Table 1). A bootstrap analysis was applied to observe how the DNA methylation patterns of the distinct datasets would correlate. The correlation between the DNA methylation values of the 500 randomly selected gene promoters was high in all scenarios, even when comparing TCGA data to control melanocytes (Table 1), similar to our CM group. This finding suggests that, in general, the pattern of gene promoter methylation in all samples (TCGA tumours, our primary CM cohort, and control melanocytes) is very similar, reinforcing the specificity of the identified signature of 514 DMGs for melanomas.
The signature of 514 DMGs was also tested on HM450K data from 259 lymph node metastases and 48 distant metastases of CMs deposited in TCGA. The correlation of gene promoter methylation levels between our cohort of CMs and TCGA metastasis sets was higher than the correlation obtained using control melanocytes (Fig. 1).

Correlation of gene promoter methylation levels between of CMs cohort and TCGA metastasis sets. Correlation of promoter DNA methylation levels of 514 differentially methylated genes between 20 primary cutaneous melanoma samples, control melanocytes and metastatic tissues (both distant and lymph node metastases retrieved from TCGA dataset) (p-value <2.2e-16)
Discussion
Driver mutations in melanomas are believed to promote evolutionary advantages for mutated cells in the tumour microenvironment, as has been described in other cancers [12], resulting in clonal expansion. Additionally, it is known that tumour progression is affected by stroma and nearby cells; specially in skin cancer, the keratinocytes accumulate alterations and are associated with various stages of melanoma development [13, 14]. Therefore, we are aware that key aspects of the melanoma-stroma interplay were not addressed by our study design, based on the comparison with data from cultured melanocytes.
Even though most driver genes do not encode proteins involved in the epigenetic machinery, we could reason that cells with distinct sets of driver mutations likely behave and evolve differently, resulting in (or as a result of) different epigenetic landscapes. Hou and colleagues observed that BRAFV600E knockdown altered the DNA methylation landscape of cells in a melanoma cell line; therefore, specific suppression of the previously over-activated MAP kinase pathway resulted in the differential activity of the epigenetic machinery in those cells [6]. This group later published another work showing a set of epigenetic genes that are up- and down-regulated upon suppression of BRAFV600E signalling in melanoma cell lines [15].
However, when we combine both the mutational and epigenetic landscapes of primary melanomas, it can be erroneous to attribute an epigenetic signature to a particular driver mutation primarily because tumours often accumulate several driver alterations [16]. Beyond a possible direct relation between acquiring a given mutation and further disturbance of the epigenetic landscape, a broad epigenetic signature could be present, which may have been generated before acquisition of the full spectrum of mutations, such as a root epigenetic programme for the oncogenesis of each particular cell type. Supporting our hypothesis, a recent work from TCGA reported a CpG island methylator phenotype (CIMP) that was not driven by events that led to the distinct genotypic subtypes [17]. The only strong association with the CIMP cluster was the presence of mutations in IDH1 and ARID2 genes, both of which are chromatin-remodelling genes.
A common group of 514 genes emerged after overlapping the sets of DMGs observed in melanomas carrying different sets of driver mutations. Interestingly, although disclosed in a small cohort, this epigenetic signature was validated in a larger independent dataset of malignant melanomas from TCGA. Thomas and colleagues used a similar assay to investigate DNA methylation differences between CM and benign nevi [18]. The authors found 145 genes with significant DNA methylation differences between the two groups. When comparing our 514 DMG signature with Thomas list, we found 10 genes in common (ALOX12, ARHGDIB, COL1A2, EMR3, FRZB, GNMT, HLA-DPB1, HOXA11, OSM, RARA), supporting our findings. Lauss and colleagues performed an integrative analysis between DNA methylation and gene expression in melanomas, and found 1119 genes that presented a negative correlation between DNA methylation and gene expression [19]. Interestingly, 65 of these genes were also present in our DMG signature, and were enriched in pathways related to tryptophan metabolism, and the Jak-STAT signalling pathway. The association of DNA methylation events with pathologic features used for the AJCC melanoma staging made by Thomas and colleagues, [20] revealed a short list of 17 differentially methylated genes. Two of these, HOXA9 and MMP14, associated with tumour thickness and mitotic rate, respectively, were also present in our DMG signature.
Some of the aberrantly methylated genes have already been determined to be related to melanomas, such as PRAME, an antigen that is preferentially expressed in human melanomas [21], as well as AIM2 (absent in melanoma 2 [22]), S100A4 [23], and TRPM1, which is inversely correlated with melanoma aggressiveness and is used as a prognostic marker for melanoma metastasis [24].
Conclusions
With these exciting results, we hope to highlight the contribution of epigenetic deregulation to melanoma development. This signature of aberrant methylation in promoter CpG sites of 514 genes can also be tested in precursor lesions, such as dysplastic nevi, as well as other types of skin and solid tumours, which may open a new avenue for early diagnosis. In fact, Gao and colleagues developed an algorithm that incorporates the promoter DNA methylation status of five genes (three of them present in our epigenetic signature – CLDN11, PPP1R3C and GNMT), that can distinguish melanoma from dysplastic nevus [25].
References
Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi:10.1101/gad.947102.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi:10.3322/caac.21254.
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–16. doi:10.1056/NEJMoa1103782.
Tanemura A, Terando AM, Sim M-S, van Hoesel AQ, de Maat MFG, Morton DL, et al. CpG island methylator phenotype predicts progression of malignant melanoma. Clin Cancer Res. 2009;15:1801–7. doi:10.1158/1078-0432.CCR-08-1361.
Mori T, O’Day SJ, Umetani N, Martinez SR, Kitago M, Koyanagi K, et al. Predictive utility of circulating methylated DNA in serum of melanoma patients receiving biochemotherapy. J Clin Oncol. 2005;23:9351–8. doi:10.1200/JCO.2005.02.9876.
Hou P, Liu D, Dong J, Xing M. The BRAF(V600E) causes widespread alterations in gene methylation in the genome of melanoma cells. Cell Cycle. 2012;11:286–95. doi:10.4161/cc.11.2.18707.
Pramio DT, Pennacchi PC, Maria-Engler SS, Campos AHJFM, Duprat JP, Carraro DM, et al. LINE-1 hypomethylation and mutational status in cutaneous melanomas. J Investig Med. 2016;64:899–904. doi:10.1136/jim-2016-000066.
Pennacchi PC, de Almeida MES, Gomes OLA, Faião-Flores F, de Araújo Crepaldi MC, Dos Santos MF, et al. Glycated reconstructed human skin as a platform to study the pathogenesis of skin aging. Tissue Eng Part A. 2015;21:2417–25. doi:10.1089/ten.TEA.2015.0009.
Brohem CA, da Silva Cardeal LB, Tiago M, Soengas MS, de Moraes Barros SB, Maria-Engler SS. Artificial skin in perspective: concepts and applications. Pigment Cell Melanoma Res. 2011;24:35–50. doi:10.1111/j.1755-148X.2010.00786.x.
Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, Gomez-Cabrero D, et al. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics. 2013;29:189–96. doi:10.1093/bioinformatics/bts680.
Wang J, Duncan D, Shi Z, Zhang B. WEB-based GEne SeT AnaLysis toolkit (WebGestalt): update 2013. Nucleic Acids Res. 2013;41:W77–83. doi:10.1093/nar/gkt439.
Nik-Zainal S, Van Loo P, Wedge DC, Alexandrov LB, Greenman CD, Lau KW, et al. The life history of 21 breast cancers. Cell. 2012;149:994–1007. doi:10.1016/j.cell.2012.04.023.
Ferrarelli LK. Melanoma cells talk to keratinocytes. Science. 2017;355(6330):1169–71. doi:10.1126/science.355.6330.1169-q.
Ostalecki C, Lee JH, Dindorf J, Collenburg L, Schierer S, Simon B, et al. Multiepitope tissue analysis reveals SPPL3-mediated ADAM10 activation as a key step in the transformation of melanocytes. Sci Signal. 2017;10(470) doi:10.1126/scisignal.aai8288.
Liu D, Liu X, Xing M. Epigenetic genes regulated by the BRAFV600E signaling are associated with alterations in the methylation and expression of tumor suppressor genes and patient survival in melanoma. Biochem Biophys Res Commun. 2012;425:45–50. doi:10.1016/j.bbrc.2012.07.046.
Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat J-P, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–63. doi:10.1016/j.cell.2012.06.024.
Akbani R, Akdemir KC, Aksoy BA, Albert M, Ally A, Amin SB, et al. Genomic classification of cutaneous melanoma. Cell. 2015;161:1681–96. doi:10.1016/j.cell.2015.05.044.
Conway K, Edmiston SN, Khondker ZS, Groben PA, Zhou X, Chu H, et al. DNA-methylation profiling distinguishes malignant melanomas from benign nevi. Pigment Cell Melanoma Res. 2011;24:352–60. doi:10.1111/j.1755-148X.2011.00828.x.
Lauss M, Haq R, Cirenajwis H, Phung B, Harbst K, Staaf J, et al. Genome-wide DNA methylation analysis in melanoma reveals the importance of CpG methylation in MITF regulation HHS public access. J Invest Dermatol. 2015;135:1820–8. doi:10.1038/jid.2015.61.
Thomas NE. Slater N a. Zhou X, Kuan P-F, Groben P a, et al. DNA-methylation Profiles in Primary Cutaneous Melanomas are Associated with Clinically Significant Pathologic Features. Pigment Cell Melanoma Res: Edmiston SN; 2014. doi:10.1111/pcmr.12289.
Ikeda H, Lethé B, Lehmann F, van Baren N, Baurain JF, de Smet C, et al. Characterization of an antigen that is recognized on a melanoma showing partial HLA loss by CTL expressing an NK inhibitory receptor. Immunity. 1997;6:199–208.
DeYoung KL, Ray ME, Su YA, Anzick SL, Johnstone RW, Trapani JA, et al. Cloning a novel member of the human interferon-inducible gene family associated with control of tumorigenicity in a model of human melanoma. Oncogene. 1997;15:453–7. doi:10.1038/sj.onc.1201206.
Haase-Kohn C, Wolf S, Lenk J, Pietzsch J. Copper-mediated cross-linking of S100A4, but not of S100A2, results in proinflammatory effects in melanoma cells. Biochem Biophys Res Commun. 2011;413:494–8. doi:10.1016/j.bbrc.2011.08.132.
Miller AJ, Du J, Rowan S, Hershey CL, Widlund HR, Fisher DE. Transcriptional regulation of the melanoma prognostic marker melastatin (TRPM1) by MITF in melanocytes and melanoma. Cancer Res. 2004;64:509–16.
Gao L, van den Hurk K, Moerkerk PTM, Goeman JJ, Beck S, Gruis NA, et al. Promoter CpG Island Hypermethylation in dysplastic nevus and melanoma: CLDN11 as an epigenetic biomarker for malignancy. J Invest Dermatol. 2014;134:2957–66. doi:10.1038/jid.2014.270.
Acknowledgments
The authors acknowledge the A.C.Camargo Cancer Center Biobank for providing patient samples.
Funding
This work was funded by FAPESP (grants 2013/10785–5 and 2013/07480–8), and CNPq (470446_2013–7).
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
ACVK conceived the study, and participated in its design and coordination; DTP designed and performed the experiments; DTP, and ACVK analyzed the data; AYK performed bioinformatics analysis; AHJFMC, JPD, and DMC assessed the clinical data and selected individuals; PCP, and SSME established primary cultures of melanocytes; DTP, and MPR wrote and edited the manuscript; ACVK critically revised the manuscript; all authors have read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All tissue samples were obtained from the AC Camargo Cancer Center Biobank (ACCCC, Sao Paulo, Brazil). This project was approved by the ethics committee of the AC Camargo Cancer Center institution (CEP ACCCC 1765/13).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1: Figure S1.
Description of clinical data and mutational profiles of 20 primary cutaneous melanomas (previously described in [7]). Each case is depicted in a different column. The mutational profile of the CMs is depicted in the upper panel, whereas the compilation of clinical data are shown in the lower panel. (XLSX 13 kb)
Additional file 2: Table S1.
Number of differentially methylated genes in the comparison of cutaneous melanomas carrying distinct driver mutations with the group of control melanocytes. ΔM refers to the mathematical conversion of beta values extracted from HM450K, which reflect the absolute methylation level for a given probe on the microarray platform. *Activating MAPK: any tumour with mutations in BRAF, NRAS and KIT. (XLSX 10 kb)
Additional file 3: Table S2.
List of the 514 differentially methylated gene promoters detected in the primary CMs compared to control melanocytes. (XLSX 15 kb)
Additional file 4: Table S3.
Gene Ontology analysis using the common set of 514 differentially methylated genes detected in primary cutaneous melanomas compared to control melanocytes. *We used WebGestalt software, which was previously used to classify the 514 DMGs in hypermethylated and hypomethylated sites in tumours. (XLSX 10 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Pramio, D.T., Kashiwabara, A.Y., Pennacchi, P.C. et al. Epigenetic signature of differentially methylated genes in cutaneous melanoma. Appl Cancer Res 37, 34 (2017). https://doi.org/10.1186/s41241-017-0041-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41241-017-0041-9