
Abstract
Throughout adult life, most lineages of blood cells, including immune cells, are generated from hematopoietic stem cells (HSCs) in the bone marrow. HSCs are thought to require special microenvironments, termed niches, for their maintenance in the bone marrow; however, the identity of the HSC cellular niche has been a subject of long-standing debate. Although diverse candidates have been proposed so far, accumulated studies demonstrate that the bone marrow-specific population of fibroblastic reticular cells with long processes, termed CXC chemokine ligand 12-abundant reticular cells (which overlap strongly with leptin receptor-expressing cells), termed CAR/LepR+ cells, are the pivotal cellular component of niches for HSCs and lymphoid progenitors. Sinusoidal endothelial cells (ECs) are also important for hematopoietic homeostasis and regeneration. Hematopoiesis is altered dynamically by various stimuli such as inflammation, infection, and leukemia, all of which affect cellular niches and alter their function. Therefore, it is important to consider situations in which stimuli affect HSCs, either via direct interaction or indirectly via the hematopoietic niches. In this review, the dynamics of cellular niches in the steady state and disease are described, with a focus on CAR/LepR+ cells and ECs.
Similar content being viewed by others

Background
Most types of blood cells, including immune cells, are generated from hematopoietic stem cells (HSCs) in the bone marrow. Early studies show that bone marrow-derived stromal cells support granulopoiesis and B lymphopoiesis when cultured for a long period, suggesting the existence of cellular niches for HSCs in the bone marrow [1, 2]. However, the identity of the niche has long been unclear. Bone-lining osteoblasts were the first cells proposed to be cellular niches for HSC [3, 4]. Then, diverse candidate cells for HSC niches, such as sinusoidal endothelial cells (ECs) [5], CXC chemokine ligand 12 (CXCL12)-abundant reticular (CAR) cells [6, 7], GFAP+ Schwann cells [8], macrophages [9, 10], Tregs [11], NG2+ periarteriolar cells [12, 13], and megakaryocytes [14, 15], had been reported so far. Simultaneously, the importance of these candidates had been confirmed or doubted by many studies [16,17,18]. For example, with several lines of negative evidence [19,20,21], osteoblasts have now lost their basis as HSC niches.
Accumulated series of evidence demonstrate that CAR cells, which overlap strongly with leptin receptor (LepR)-expressing cells, are the major cellular component of niches for hematopoietic stem and progenitor cells (HSPCs) [6, 7, 22,23,24]. Sinusoidal endothelial cells are also important for hematopoietic homeostasis and regeneration [22, 25]. High-throughput gene expression analysis at the single-cell level, which has developed rapidly in recent years, allows comprehensive analysis of hematopoietic cells and their cellular niches in the bone marrow. Furthermore, it has been clear that infection, inflammation, and leukemia lead to dynamic changes in the cellularity and properties of these cellular niches, as well as the HSPCs themselves.
CAR/LepR+ cells
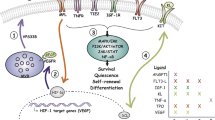
Stem cell factor (SCF), CXCL12, and thrombopoietin (TPO) are essential cytokines involved in maintenance of HSCs. Mice deficient in any of these genes show a marked reduction in the number of HSCs in the bone marrow [26,27,28,29,30]. These facts suggest that cells producing these cytokines are an important component of the HSC niche. However, of these three cytokines, TPO is the exception because it is synthesized mainly outside the bone marrow and acts as a hormone. TPO is expressed by hepatocytes, and its deletion from hepatocytes depletes HSCs from the bone marrow [31, 32].
To identify cellular niches for HSCs in the bone marrow, Sugiyama et al. generated mice in which a green fluorescent protein (GFP) reporter gene was knocked into the CXCL12 locus (CXCL12-GFP mice); they then identified a population of reticular cells with long processes that express much higher levels of CXCL12 than any other types of cells in the bone marrow and other organs. These cells were named CXCL12-abundant reticular (CAR) cells [6]. Furthermore, short-term ablation of CAR cells in vivo, using mice in which a transgene encoding the diphtheria toxin receptor (DTR) was knocked into the CXCL12 locus (CXCL12-DTR mice), led to a marked reduction of CXCL12 and SCF in the bone marrow, indicating that CAR cells are the major producer of CXCL12 and SCF in the bone marrow [7]. Subsequently, Ding et al. generated mice in which GFP was knocked into the SCF locus (SCF-GFP mice), and showed that these cells express much higher levels of SCF (overlapping strongly with CAR cells); in addition, most of these cells express much higher levels of LepR than other types of bone marrow cell [22]. Therefore, these cells will be referred to as CAR/LepR+ cells in the following. Recent single-cell RNA sequencing studies also revealed that CAR/LepR+ cells are a distinct cell population that expresses high levels of SCF and CXCL12 [33,34,35].
Méndez-Ferrer et al. reported Nestin-GFP positive cells as HSC niches by using Nestin-GFP transgenic mice, in which GFP is expressed under the control of the neural-specific regulatory elements of Nestin gene [36]. It is now known that Nestin-GFPhigh cells and Nestin-GFPlow cells are distinct cells; Nestin-GFPlow cells strongly overlap with CAR/LepR+ cells, and Nes-GFPhigh cells overlap with Myh11+ NG2+ cells in the periarterial area [13, 36, 37]. It is noted that adult CAR/LepR+ cells or Nestin-GFPlow cells do not express endogenous Nestin [12, 37]. In addition, Nestin-Cre or NG2-Cre label both Nestin-GFPhigh and Nestin-GFPlow (or CAR/LepR+) cells, but NG2-CreER and Myh11-CreERT2 label peri-arterial NG2+ cells but not CAR/LepR+ cells [13, 22, 37]. Some osteoblastic lineage Cre-lines, such as Sp7-Cre, Ocn-Cre, and Dmp1-Cre, also label varying parts of CAR/LepR+ cells [38, 39]. Adipoq-Cre labels adipocytes and probably most CAR/LepR+ cells, while Adipoq-CreER labels adipocytes and a few CAR/LepR+ cells [40, 41].
Selective ablation of CAR/LepR+ cells using CXCL12-DTR mice [7] or mice in which DTR is expressed by LepR+ cells [37] leads to a decrease in the number of HSPCs. Similarly, when SCF is deleted from LepR+ cells, the number of HSPCs in the bone marrow decreases markedly [22, 42]. Interleukin-7 (IL-7) is also expressed by most CAR/LepR+ cells, and its deletion from CAR/LepR+ cells leads to a severe reduction in the numbers of pro-B and pre-B cells in the bone marrow [43]. Taken together, these results indicate that CAR/LepR+ cells play a critical role in maintaining not only HSCs but also B cell progenitors.
Foxc1 was first identified as an essential transcription factor that is preferentially expressed by CAR cells in the bone marrow. When Foxc1 is conditionally deleted from CAR/LepR+ cells, the number of HSPCs is markedly reduced, accompanied by a marked increase in the number of adipocytes in the bone marrow [23]. These results indicate that Foxc1 is essential for the maintenance of niches for HSPCs and for inhibiting differentiation of CAR cells into adipocytes (Fig. 1). Another transcription factor, Ebf3, is also expressed preferentially by CAR cells. Ebf1 is expressed by B cell precursors and is critical for B cell development; however, it is expressed preferentially by CAR cells among all bone marrow non-hematopoietic cells. When both Ebf3 and Ebf1 are deleted from CAR/LepR+ cells, the number of HSPCs in the bone marrow is markedly reduced, accompanied by a marked increase of trabecular bones [24]. These results indicate that Ebf1/3 play an essential role in the maintenance of HSPC niches and the bone marrow cavity by inhibiting the differentiation of CAR cells into osteoblasts. Transcription factor Snai2 is also expressed at high levels by CAR/LepR+ cells, and Snai2 null mice (129Sv/C57BL/6 F1 background) or conditional knockout mice (Mx1-Cre; Snai2-flox) show slightly decreased HSC numbers in the bone marrow. It is argued that increased expression of osteopontin (Spp1) may be responsible for the phenotype, but precise verification using CAR/LepR+ cell-specific knockout mice would be needed to confirm this [44]. A more recent study showed that transcription factors Runx1 and Runx2 are expressed at high levels by CAR cells and that the bone marrow is markedly fibrotic and hematopoiesis is severely impaired in Runx1 and Runx2 conditional knockout mice. These results indicate that CAR cells require Runx1 or Runx2 to prevent their fibrotic conversion and to maintain HSCs and hematopoiesis in adults [45]. Elucidation of the interactions and functional links among these transcription factors will be the subject of future studies.

Essential transcription factors in CAR/LepR+ cells
Sinusoidal endothelial cells
Bone marrow venous endothelial cells, termed sinusoidal endothelial cells, are thought to be a component of the HSPC niche because they promote the maintenance of HSPCs in culture [25, 46]. Bone marrow endothelial cells express low levels of SCF and CXCL12, which are required for the maintenance and regeneration of HSPCs [7, 33,34,35]. Conditional deletion of either of these factors from endothelial cells decreases the number of HSPCs in the bone marrow [22]. However, it remains unclear whether endothelial cells synthesize these factors entirely in the bone marrow, or whether endothelial cells outside the bone marrow contribute to their production. In the fetal stage when HSCs proliferate in the liver, SCF derived from hepatic stellate cells is important, but EC-derived SCF also shows some contribution [47]. The sinusoidal endothelium might be essential for hematopoietic homeostasis due to its function as a regulated gate for differentiated blood cells, rather than as a source of hematopoietic cytokines. Furthermore, sinusoidal endothelial cells appear to play an important role in the regeneration of hematopoiesis after bone marrow destruction [25, 48].
Bone marrow endothelial cells are divided into subpopulations based on differential expression of PECAM-1 (CD31) and endomucin (Emcn). Type-H (CD31hi Emcnhi) endothelial cells are tightly coupled to osteogenesis and localize in the metaphysis and endosteum of postnatal long bone [49, 50]. Notch signaling and integrin-β1 expression by endothelial cells are required for the specificity and function of Type-H subpopulations associated with osteoprogenitors [50, 51]. However, the molecular basis of Type-L (CD31lo Emcnlo) endothelial cells, which comprise the majority of the bone marrow space during hematopoiesis, is largely unknown.
The bone marrow microenvironment at single-cell resolution
In recent years, a series of reports have undertaken comprehensive transcriptome analysis of non-hematopoietic cells in the bone marrow at the single-cell level. These studies confirmed that CAR/LepR+ cells are the major producers of hematopoietic cytokines, suggesting a minimal contribution by other non-hematopoietic cells such as endothelial cells, osteoblasts, and nerve cells [33,34,35]. It is also suggested that the CAR/LepR+ cell cluster, which shows uniform expression of key niche factor genes such as Cxcl12, Kitl, Foxc1, and Ebf3, may be divided into several subclusters based on the expression of cell differentiation marker genes. Although these subclusters are likely to reflect their cellular origin or some bias in differentiation potential, their boundaries are ambiguous. Their specific functions and potentials should be fully validated by subcluster-specific depletion and/or tracing. For example, CAR/LepR+ subpopulations that produce high levels of osteolectin localize near the endosteum and periarterial areas, and they are suggested to be particularly supportive of B-cell development [52]. Further studies are needed to identify differences in cell lineages, as well as the specific functions of these subpopulations of CAR/LepR+ cells.
Furthermore, these comprehensive analyses revealed dynamic changes in the proportions of cell clusters, as well as gene expression, in bone marrow non-hematopoietic cell populations during stresses such as 5-FU administration [33] and leukemia [34]. In the future, these analyses at single-cell resolution will provide new findings that go beyond confirming the results of conventional methods. However, it should be noted that single-cell analysis may not include all cells due to technical limitations. For example, macrophage populations in the bone marrow are difficult to isolate without enzymatic treatment, meaning that macrophage-derived cell fragments and mRNA can contaminate other cell populations [53].
The niche changes in response to inflammation, infection, and leukemia
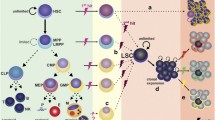
HSPCs can sense and respond to pathogen-derived molecules and inflammatory cytokines directly [54, 55]. On the other hand, the bone marrow microenvironment also alters its niche function by sensing stimuli such as pathogens, inflammatory cytokines, and leukemic cells [16, 56, 57]. Therefore, it is necessary to consider cases in which those stimuli affect HSPCs directly, and when they affect HSPCs indirectly via the hematopoietic niches (Fig. 2).

Two pathways that impair normal hematopoiesis upon inflammation, infection, or leukemia
Endothelial cells express several pattern recognition receptors (PRRs) such as toll-like receptors (TLRs), which sense pathogens, and activate HSPCs to proliferate and accelerate myelopoiesis (emergency hematopoiesis). For example, expression of TLR4 and MyD88 by endothelial cells is required for neutrophil recruitment in response to lipopolysaccharides (LPS) derived from Escherichia coli, and subsequent secretion of granulocyte colony-stimulating factor (G-CSF) initiates emergency hematopoiesis [58].
Mesenchymal cell populations, including CAR/LepR+ cells, also recognize surrounding inflammatory signals that occur locally or systemically during infection and inflammation. Activation of various PRRs leads to the production of additional factors and alters the expression of cytokines and chemokines in cellular niches. Studies using models of acute viral infection suggest that mesenchymal cells are stimulated by interferon-γ to secrete interleukin-6 (IL-6), leading to the expansion of myeloid progenitors and mature myeloid cells [59]. Similarly, in chronic repetitive LPS-induced inflammation, CAR cells are the main producers of IL-6, which is important for sustained myeloid production [60]. Furthermore, upregulation of G-CSF during infection is thought to promote myeloid hematopoiesis and suppress CXCL12 production by CAR cells, resulting in the mobilization of HSPCs to the circulation [61]. In addition, CAR/LepR+ cells increase the number of circulating monocytes by upregulating the expression of the migratory chemokine CCL2, which is important for efficient clearance of Listeria monocytogenes infection [62].
Radiation and chemotherapies, such as 5-fluorouracil (5-FU), cause myeloablation, an increase in adipocytes, and damage to the vascular network [33, 41, 48]. The mechanisms underlying endothelial cell regeneration after injury are at least partially related to VEGFR2 signaling [48]. Furthermore, the transplantation of granulocytes into irradiated mice enhances endothelial cell regeneration via TNF-α signaling [63]. These studies on dynamics, as well as studies that identify essential signals for endothelial cells and CAR/LepR+ cells during the regeneration process, will have important implications for clinical applications.
Many studies report changes in the bone marrow environment during leukemia. It has become clear that leukemic cells can alter the hematopoietic niche in a variety of ways and disrupt homeostasis of hematopoietic cells. Studies in a mouse model of myeloproliferative neoplasms such as chronic myeloid leukemia (CML) revealed that malignant cells transform osteoblast lineage cells (maybe including CAR/LepR+ cells) into inflammatory myelofibrotic cells, supporting malignant cells at the expense of normal hematopoiesis [64]. In addition, it has been reported that a major cause of hematopoietic defect is IL-6 produced by CML cells [65], which impacts both HSPC and their cellular niches. Indeed, HSPC niche function is impaired including decreased expression of CXCL12 in CAR cells in the CML mice [66, 67]. These results were confirmed in recently identified human CAR cells from CML patients [68]. Furthermore, thymidine kinase inhibitor (TKI)-resistant leukemic stem cells (LSCs) in CML are maintained in the bone marrow in a CXCL12-dependent manner, indicating that loss of CXCL12 inhibits maintenance of LSC quiescence and makes them sensitive to TKIs [67]. Primary myelofibrosis (PMF) is associated with bone marrow fibrosis; mouse models show that transforming growth factor-beta (TGF-β) and platelet-derived growth factors (PDGFs) are involved in fibrosis and that the source of myofibroblasts is CAR/LepR+ cells [69]. It has also been shown that altered cellular niches produce alarmins such as S100A8/A9 that accelerate fibrosis in PMF model mice [70]. Interestingly, in myelodysplastic syndrome (MDS) model mice, the involvement of extracellular vesicles containing microRNAs has been suggested as leukemic cell-derived factors that suppress the niche function for normal hematopoiesis [71].
Thus, the bone marrow hematopoietic environment is thought to be altered differently by inflammation, infection, or leukemia. These changes lead to differences in hematopoietic activity and/or resistance to therapy. In the steady state, CAR/LepR+ cells act as central cellular niches, but this is not always the case in disease. Certain malignant cells that actively impair hematopoiesis may emerge during disease states. To understand precisely the heterogeneous hematopoietic environment in disease conditions, comprehensive single-cell level analysis can be a powerful tool [33, 34, 70]. These analyses have been expected to reveal the key cellular components controlling each disease as therapeutic targets. Further studies will be needed to reveal the actual effector cells involved and the essential molecular mechanisms underlying each phenomenon.
Conclusions
There is accumulating evidence for a hematopoietic microenvironment in the bone marrow. In particular, comprehensive analysis at the single-cell level has confirmed that CAR/LepR+ cells play a pivotal role in the HSPC niche as they produce the greatest amounts of cytokines that are essential for hematopoiesis.
Inflammation, infection, and leukemia have a significant effect on CAR cells and endothelial cells, as well as HSPCs, leading to altered functions. Depending on the situation, the actual effector cells and the molecular mechanisms might be diverse, and many are not fully understood. In addition, we do not really know how long changes in the niche persist, or how the cellular niches and normal hematopoietic cells regenerate. Because various hematopoietic and non-hematopoietic cells can be effectors and/or mediators, the dynamics are quite complex. Thus, a comprehensive view of all cells in the bone marrow is needed. It is important to identify key cell types and key signaling pathways that function during changes in the bone marrow because they may be clinical targets for controlling hematopoietic homeostasis.
Availability of data and materials
Not applicable.
Abbreviations
- HSC:
-
Hematopoietic stem cell
- CAR:
-
CXCL12-abundant reticular
- EC:
-
Endothelial cell
- HSPC:
-
Hematopoietic stem/progenitor cell
- GFAP:
-
Glial fibrillary acidic protein
- SCF:
-
Stem cell factor
- TPO:
-
Thrombopoietin
- GFP:
-
Green fluorescent protein
- DTR:
-
Diphtheria toxin receptor
- PRR:
-
Pattern recognition receptor
- LCMV:
-
Lymphocytic choriomeningitis virus
- TGF-β:
-
Transforming growth factor-beta
- PDGF:
-
Platelet-derived growth factor
- CML:
-
Chronic myeloid leukemia
- TKI:
-
Thymidine kinase inhibitor
- LSC:
-
Leukemic stem cell
- PMF:
-
Primary myelofibrosis
References
Dexter TM, Allen TD, Lajtha LG. Conditions controlling the proliferation of haemopoietic stem cells in vitro. J Cell Physiol. 1977;91:335–44.
Whitlock CA, Witte ON. Long-term culture of B lymphocytes and their precursors from murine bone marrow. Proc Natl Acad Sci U S A. 1982;79(11 I):3608–12.
Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425:841–6.
Zhang J, Niu C, Ye L, Huang H, He X, Tong WG, et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425:836–41.
Kiel MJ, Yilmaz ÖH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121:1109–21.
Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25:977–88.
Omatsu Y, Sugiyama T, Kohara H, Kondoh G, Fujii N, Kohno K, et al. The essential functions of adipo-osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity. 2010;33:387–99.
Yamazaki S, Ema H, Karlsson G, Yamaguchi T, Miyoshi H, Shioda S, et al. Nonmyelinating Schwann cells maintain hematopoietic stem cell hibernation in the bone marrow niche. Cell. 2011;147:1146–58.
Chow A, Lucas D, Hidalgo A, Méndez-Ferrer S, Hashimoto D, Scheiermann C, et al. Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J Exp Med. 2011;208:761–71.
Ludin A, Itkin T, Gur-Cohen S, Mildner A, Shezen E, Golan K, et al. Monocytes-macrophages that express α-smooth muscle actin preserve primitive hematopoietic cells in the bone marrow. Nat Immunol. 2012;13:1072–82.
Fujisaki J, Wu J, Carlson AL, Silberstein L, Putheti P, Larocca R, et al. In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature. 2011;474:216–9.
Kunisaki Y, Bruns I, Scheiermann C, Ahmed J, Pinho S, Zhang D, et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature. 2013;502:637–43.
Asada N, Kunisaki Y, Pierce H, Wang Z, Fernandez NF, Birbrair A, et al. Differential cytokine contributions of perivascular haematopoietic stem cell niches. Nat Cell Biol. 2017;19:214–23.
Bruns I, Lucas D, Pinho S, Ahmed J, Lambert MP, Kunisaki Y, et al. Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat Med. 2014;20:1315–20.
Zhao M, Perry JM, Marshall H, Venkatraman A, Qian P, He XC, et al. Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nat Med. 2014;20:1321–6.
Comazzetto S, Shen B, Morrison SJ. Niches that regulate stem cells and hematopoiesis in adult bone marrow. Dev Cell. 2021;56:1848–60.
Omatsu Y, Nagasawa T. Identification of microenvironmental niches for hematopoietic stem cells and lymphoid progenitors-bone marrow fibroblastic reticular cells with salient features. Int Immunol. 2021;33:821–6.
Omatsu Y, Higaki K, Nagasawa T. Cellular niches for hematopoietic stem cells and lympho-hematopoiesis in bone marrow during homeostasis and blood cancers. Curr Top Microbiol Immunol. 2021;434:33–54.
Kiel MJ, Radice GL, Morrison SJ. Lack of evidence that hematopoietic stem cells depend on N-cadherin-mediated adhesion to osteoblasts for their maintenance. Cell Stem Cell. 2007;1:204–17.
Bowers M, Zhang B, Ho Y, Agarwal P, Chen CC, Bhatia R. Osteoblast ablation reduces normal long-term hematopoietic stem cell self-renewal but accelerates leukemia development. Blood. 2015;125:2678–88.
Yu VWC, Saez B, Cook C, Lotinun S, Pardo-Saganta A, Wang Y-H, et al. Specific bone cells produce DLL4 to generate thymus-seeding progenitors from bone marrow. J Exp Med. 2015;212:759–74.
Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature. 2012;481:457–62.
Omatsu Y, Seike M, Sugiyama T, Kume T, Nagasawa T. Foxc1 is a critical regulator of haematopoietic stem/progenitor cell niche formation. Nature. 2014;508:536–40.
Seike M, Omatsu Y, Watanabe H, Kondoh G, Nagasawa T. Stem cell niche-specific Ebf3 maintains the bone marrow cavity. Genes Dev. 2018;32:359–72.
Butler JM, Nolan DJ, Vertes EL, Varnum-Finney B, Kobayashi H, Hooper AT, et al. Endothelial cells are essential for the self-renewal and repopulation of Notch-dependent hematopoietic stem cells. Cell Stem Cell. 2010;6:251–64.
Barker JE. SI/SI(d) hematopoietic progenitors are deficient in situ. Exp Hematol. 1994;22:174–7.
Nagasawa T, Hirota S, Tachibana K, Takakura N, Nishikawa SI, Kitamura Y, et al. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature. 1996;382:635–8.
Ara T, Tokoyoda K, Sugiyama T, Egawa T, Kawabata K, Nagasawa T. Long-term hematopoietic stem cells require stromal cell-derived factor-1 for colonizing bone marrow during ontogeny. Immunity. 2003;19:257–67.
Egawa T, Kawabata K, Kawamoto H, Amada K, Okamoto R, Fujii N, et al. The earliest stages of B cell development require a chemokine stromal cell-derived factor/pre-B cell growth-stimulating factor. Immunity. 2001;15:323–34.
de Sauvage FJ, Carver-Moore K, Luoh SM, Ryan A, Dowd M, Eaton DL, et al. Physiological regulation of early and late stages of megakaryocytopoiesis by thrombopoietin. J Exp Med. 1996;183:651–6.
Decker M, Leslie J, Liu Q, Ding L. Hepatic thrombopoietin is required for bone marrow hematopoietic stem cell maintenance. Science. 2018;360:106–10.
O’Neill A, Chin D, Tan D, Abdul Majeed ABB, Nakamura-Ishizu A, Suda T. Thrombopoietin maintains cell numbers of hematopoietic stem and progenitor cells with megakaryopoietic potential. Haematologica. 2021;106:1883–91.
Tikhonova AN, Dolgalev I, Hu H, Sivaraj KK, Hoxha E, Cuesta-Domínguez Á, et al. The bone marrow microenvironment at single-cell resolution. Nature. 2019;569:222–8.
Baryawno N, Przybylski D, Kowalczyk MS, Kfoury Y, Severe N, Gustafsson K, et al. A cellular taxonomy of the bone marrow stroma in homeostasis and leukemia. Cell. 2019;177:1915–1932.e16.
Baccin C, Al-Sabah J, Velten L, Helbling PM, Grünschläger F, Hernández-Malmierca P, et al. Combined single-cell and spatial transcriptomics reveal the molecular, cellular and spatial bone marrow niche organization. Nat Cell Biol. 2020;22:38–48.
Méndez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, MacArthur BD, Lira SA, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466:829–34.
Zhou BO, Yue R, Murphy MM, Peyer JG, Morrison SJ. Leptin-receptor-expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell. 2014;15:154–68.
Greenbaum A, Hsu YMS, Day RB, Schuettpelz LG, Christopher MJ, Borgerding JN, et al. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature. 2013;495:227–30.
Zhang J, Link DC. Targeting of mesenchymal stromal cells by Cre-recombinase transgenes commonly used to target osteoblast lineage cells. J Bone Miner Res. 2016;31:2001–7.
Yu W, Zhong L, Yao L, Wei Y, Gui T, Li Z, et al. Bone marrow adipogenic lineage precursors promote osteoclastogenesis in bone remodeling and pathologic bone loss. J Clin Invest. 2021;131(2):e140214. https://doi.org/10.1172/JCI140214.
Zhou BO, Yu H, Yue R, Zhao Z, Rios JJ, Naveiras O, et al. Bone marrow adipocytes promote the regeneration of stem cells and haematopoiesis by secreting SCF. Nat Cell Biol. 2017;19:891–903.
Comazzetto S, Murphy MM, Berto S, Jeffery E, Zhao Z, Morrison SJ. Restricted hematopoietic progenitors and erythropoiesis require SCF from leptin receptor+ niche cells in the bone marrow. Cell Stem Cell. 2019;24:477–486.e6.
Cordeiro Gomes A, Hara T, Lim VY, Herndler-Brandstetter D, Nevius E, Sugiyama T, et al. Hematopoietic stem cell niches produce lineage-instructive signals to control multipotent progenitor differentiation. Immunity. 2016;45:1219–31.
Wei Q, Nakahara F, Asada N, Zhang D, Gao X, Xu C, et al. Snai2 maintains bone marrow niche cells by repressing osteopontin expression. Dev Cell. 2020;53:503–513.e5.
Omatsu Y, Aiba S, Maeta T, Higaki K, Aoki K, Watanabe H, et al. Runx1 and Runx2 inhibit fibrotic conversion of cellular niches for hematopoietic stem cells. Nat Commun. 2022;13:2654.
Li W, Johnson SA, Shelley WC, Yoder MC. Hematopoietic stem cell repopulating ability can be maintained in vitro by some primary endothelial cells. Exp Hematol. 2004;32:1226–37.
Lee Y, Leslie J, Yang Y, Ding L. Hepatic stellate and endothelial cells maintain hematopoietic stem cells in the developing liver. J Exp Med. 2021;218(3):e20200882. https://doi.org/10.1084/jem.20200882.
Hooper AT, Butler JM, Nolan DJ, Kranz A, Iida K, Kobayashi M, et al. Engraftment and reconstitution of hematopoiesis is dependent on VEGFR2-mediated regeneration of sinusoidal endothelial cells. Cell Stem Cell. 2009;4:263–74.
Kusumbe AP, Ramasamy SK, Adams RH. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature. 2014;507:323–8.
Ramasamy SK, Kusumbe AP, Wang L, Adams RH. Endothelial Notch activity promotes angiogenesis and osteogenesis in bone. Nature. 2014;507:376–80.
Langen UH, Pitulescu ME, Kim JM, Enriquez-Gasca R, Sivaraj KK, Kusumbe AP, et al. Cell-matrix signals specify bone endothelial cells during developmental osteogenesis. Nat Cell Biol. 2017;19:189–201.
Shen B, Tasdogan A, Ubellacker JM, Zhang J, Nosyreva ED, Du L, et al. A mechanosensitive peri-arteriolar niche for osteogenesis and lymphopoiesis. Nature. 2021;591:438–44.
Millard SM, Heng O, Opperman KS, Sehgal A, Irvine KM, Kaur S, et al. Fragmentation of tissue-resident macrophages during isolation confounds analysis of single-cell preparations from mouse hematopoietic tissues. Cell Rep. 2021;37:110058.
Nagai Y, Garrett KP, Ohta S, Bahrun U, Kouro T, Akira S, et al. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity. 2006;24:801–12.
Zhao JL, Ma C, O’Connell RM, Mehta A, DiLoreto R, Heath JR, et al. Conversion of danger signals into cytokine signals by hematopoietic stem and progenitor cells for regulation of stress-induced hematopoiesis. Cell Stem Cell. 2014;14:445–59.
Mitroulis I, Kalafati L, Bornhäuser M, Hajishengallis G, Chavakis T. Regulation of the bone marrow niche by inflammation. Front Immunol. 2020;11:1540. https://doi.org/10.3389/fimmu.2020.01540.
Mabuchi Y, Okawara C, Méndez-Ferrer S, Akazawa C. Cellular heterogeneity of mesenchymal stem/stromal cells in the bone marrow. Front cell Dev Biol. 2021;9:689366.
Boettcher S, Gerosa RC, Radpour R, Bauer J, Ampenberger F, Heikenwalder M, et al. Endothelial cells translate pathogen signals into G-CSF-driven emergency granulopoiesis. Blood. 2014;124:1393–403.
Schürch CM, Riether C, Ochsenbein AF. Cytotoxic CD8+ T cells stimulate hematopoietic progenitors by promoting cytokine release from bone marrow mesenchymal stromal cells. Cell Stem Cell. 2014;14:460–72.
Gerosa RC, Boettcher S, Kovtonyuk LV, Hausmann A, Hardt W-D, Hidalgo J, et al. CXCL12-abundant reticular cells are the major source of IL-6 upon LPS stimulation and thereby regulate hematopoiesis. Blood Adv. 2021;5:5002–15.
Day RB, Bhattacharya D, Nagasawa T, Link DC. Granulocyte colony-stimulating factor reprograms bone marrow stromal cells to actively suppress B lymphopoiesis in mice. Blood. 2015;125:3114–7.
Shi C, Jia T, Mendez-Ferrer S, Hohl TM, Serbina NV, Lipuma L, et al. Bone marrow mesenchymal stem and progenitor cells induce monocyte emigration in response to circulating toll-like receptor ligands. Immunity. 2011;34:590–601.
Bowers E, Slaughter A, Frenette PS, Kuick R, Pello OM, Lucas D. Granulocyte-derived TNFα promotes vascular and hematopoietic regeneration in the bone marrow. Nat Med. 2018;24:95–102.
Schepers K, Pietras EM, Reynaud D, Flach J, Binnewies M, Garg T, et al. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell. 2013;13:285–99.
Welner RS, Amabile G, Bararia D, Czibere A, Yang H, Zhang H, et al. Treatment of chronic myelogenous leukemia by blocking cytokine alterations found in normal stem and progenitor cells. Cancer Cell. 2015;27:671–81.
Zhang B, Ho YW, Huang Q, Maeda T, Lin A, Lee S, et al. Altered microenvironmental regulation of leukemic and normal stem cells in chronic myelogenous leukemia. Cancer Cell. 2012;21:577–92.
Agarwal P, Isringhausen S, Li H, Paterson AJ, He J, Gomariz Á, et al. Mesenchymal niche-specific expression of Cxcl12 controls quiescence of treatment-resistant leukemia stem cells. Cell Stem Cell. 2019;24:769–784.e6.
Aoki K, Kurashige M, Ichii M, Higaki K, Sugiyama T, Kaito T, et al. Identification of CXCL12-abundant reticular cells in human adult bone marrow. Br J Haematol. 2021;193:659–68.
Decker M, Martinez-Morentin L, Wang G, Lee Y, Liu Q, Leslie J, et al. Leptin-receptor-expressing bone marrow stromal cells are myofibroblasts in primary myelofibrosis. Nat Cell Biol. 2017;19:677–88.
Leimkühler NB, Gleitz HFE, Ronghui L, Snoeren IAM, Fuchs SNR, Nagai JS, et al. Heterogeneous bone-marrow stromal progenitors drive myelofibrosis via a druggable alarmin axis. Cell Stem Cell. 2021;28:637–52.
Hayashi Y, Kawabata KC, Tanaka Y, Uehara Y, Mabuchi Y, Murakami K, et al. MDS cells impair osteolineage differentiation of MSCs via extracellular vesicles to suppress normal hematopoiesis. Cell Rep. 2022;39:110805.
Acknowledgements
Not applicable.
Funding
This work was partially supported by the Japan Society for the Promotion of Science KAKENHI (22H05064 and 22H02850).
Author information
Authors and Affiliations
Contributions
The author read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The author declares no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Omatsu, Y. Cellular niches for hematopoietic stem cells in bone marrow under normal and malignant conditions. Inflamm Regener 43, 15 (2023). https://doi.org/10.1186/s41232-023-00267-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41232-023-00267-5