
Abstract
Pluripotent stem cells (PSCs) exhibit promising application in regenerative therapy, drug discovery, and disease modeling. While several protocols for differentiating somatic cells from PSCs exist, their use is limited by contamination of residual undifferentiated PSCs and immaturity of differentiated somatic cells.
The metabolism of PSCs differs greatly from that of somatic cells, and a distinct feature is required to sustain the distinct properties of PSCs. To date, several studies have reported on the importance of metabolism in PSCs and their derivative cells. Here, we detail advancements in the field, with a focus on cardiac regenerative therapy.
Similar content being viewed by others


Background
Human PSCs (hPSCs) are capable of self-renewal, proliferation, and differentiation into cells of the three germ layers, and show promising application in drug discovery, disease modeling, and regenerative therapy for diseases resistant to conventional medical therapies. Effective regenerative therapy is contingent on certain considerations, including the establishment of human-induced PSCs (hiPSCs) from somatic cells, expansion of hiPSCs, cell differentiation, removal of residual undifferentiated PSCs, and maturation of differentiated cells. Utilizing the metabolic features of PSCs and their derivative cells is a promising approach to address these considerations, as metabolic intervention is typically a cost-effective and relatively simple solution. To date, numerous studies have demonstrated close relationships between metabolism and cell phenotype, with metabolism and phenotype often affecting one another. This review details the current metabolic understanding of hPSCs and their derivatives, and compares them to mouse PSCs (mPSCs) and cancer cells to clarify similarities and differences among these cells.
Metabolism of undifferentiated hPSCs
Glucose
Rapidly proliferating cells, such as cancer cells, show activated glycolysis even under sufficient oxygen. This phenomenon observed in cancer cells was described by Otto Warburg nearly a century ago, and was thus named the Warburg effect [1]. A possible advantage of the Warburg effect is that it produces sufficient biomass to enable rapid proliferation, that is, production of nucleotides, nonessential amino acids, and lipids. A similar phenomenon is observed in PSCs, which show rapid proliferation similar to cancer cells. This was first described in mPSCs, and later in hPSCs [2, 3] (Fig. 1). Concordant with this report, during reprogramming from somatic cells to hiPSCs, metabolic profiles dramatically change from an oxidative state to a glycolytic state [4]. Studies comparing the gene expression profiles of hPSCs and somatic cells have demonstrated an upregulation of glycolytic genes, including hexokinase 2, with concomitant downregulation of pyruvate dehydrogenase, in hPSCs, which limits entry of glucose-derived pyruvate to the tricarboxylic (TCA) cycle [3]. Interestingly, hexokinase 2 and pyruvate kinase M2 are regulated by Oct4, and their overexpression limits differentiation in mPSCs [5]. Moreover, pyruvate kinase M2 positively upregulates the expression of Oct4 through its regulator activity, suggesting positive feedback in pluripotency and active glycolysis in mPSCs [6].

Metabolic features of hPSCs. hPSCs consume glucose for glycolysis, nucleotide synthesis, nonessential amino acid synthesis, and hexosamine biosynthesis. The hexosamine pathway produces UDP-GlcNAc, which serves as a substrate for protein O-GlcNAcylation. The entry of pyruvate into the TCA cycle is inhibited by reduced activity of PDH by PDK. Pyruvate is reduced to lactate to replenish nicotinamide adenine dinucleotide (NAD+) to maintain glycolytic activity. Pyruvate, which enters the mitochondrial matrix, is converted to acetyl-CoA and is utilized for citrate production. In hPSCs, the TCA cycle enzyme aconitase 2 is downregulated and conversion into αKG is limited. Citrate transported to the cytosol is converted back to acetyl-CoA, which is used for histone acetylation, de novo fatty acid synthesis, and hexosamine biosynthetic pathway. Fatty acids are utilized for various complex lipids, including glycosphingolipids. To replenish the latter part of the TCA cycle (i.e., αKG to oxaloacetate), hPSCs consume glutamine. Glutamine also plays a key role in glutathione synthesis, the hexosamine biosynthetic pathway, and the serine synthesis pathway. α-KG is required for histone and DNA demethylation. mPSCs consume threonine to produce SAM, but because threonine dehydrogenase is a pseudogene in hPSCs, hPSCs consume methionine to produce SAM. SAM is used for histone methylation. αKG, α-ketoglutarate; ACO2, aconitase 2; F6P, fructose 6-phosphate; FASN, fatty acid synthase; G6P, glucose 6-phosphate; Gln, glutamine; Glu, glutamate; Glucosamine-6-P, glucosamine-6-phosphate; Gly, glycine; GSH, glutathione; hCys, homocysteine; LDH, lactate dehydrogenase; Met, methionine; meTHF, 5,10-methylenetetrahydrofolate; mTHF, 5-methyltetrahydrofolate; PDH, pyruvate dehydrogenase; PDK, pyruvate dehydrogenase kinase; PKM2, pyruvate kinase M2; PSAT1, phosphoserine aminotransferase 1; R5P, ribose 5-phosphate; SAH, S-adenosyl homocysteine; SAM, S-adenosyl methionine; Ser, serine; TCA cycle, tricarboxylic acid cycle; THF, tetrahydrofolic acid; UDP-GlcNAc, uridine diphosphate N-acetylglucosamine
Reprogramming of mouse embryonic fibroblasts (MEFs) into mouse induced PSCs (miPSCs) results in the transformation of structurally (and functionally) mature mitochondria into immature mitochondria, with a concomitant increase in expression of glycolytic genes, consumption of glucose, and production of lactate. Of note, the rise in glycolytic enzymes precedes that of pluripotent markers [7]. While inhibition of glycolysis by 2-deoxy-d-glucose limits reprogramming efficiency, stimulation of glycolysis by high glucose improves it [7]. In addition, hypoxia-inducible factor 1-alpha (HIF1α) and hypoxia-inducible factor 2-alpha (HIF2α) are upregulated in the early stages of reprogramming in both hiPSCs and miPSCs [8]. HIF1α upregulates pyruvate dehydrogenase kinase, which inhibits pyruvate dehydrogenase activity, preventing the entry of pyruvate into the TCA cycle and activation of glycolysis [9].
As noted above, glycolysis is upregulated by reprogramming into induced PSCs (iPSCs). However, transient upregulation of estrogen-related receptor alpha/gamma (ERRα/γ) and their co-activators, peroxisome proliferator-activated receptor gamma coactivator 1-alpha/beta (PGC-1α/β) with a concomitant transient burst in oxidative phosphorylation (OXPHOS) are observed during reprogramming of hiPSCs and miPSCs [10].
Hexosamine
The glycolytic intermediate metabolite fructose-6-phosphate is channeled to the hexosamine biosynthetic pathway (HBP) via glutamine-fructose-6-pshophate aminotransferase 1, the first and rate-limiting enzyme of HBP. Uridine diphosphate N-acetylglucosamine (UDP-GlcNAc) is produced by the HBP from glucose, glutamine, acetyl-CoA, and uridine diphosphate, and hence serves as a nutrient sensor for these metabolites by acting as a substrate for O-GlcNAcylation, one of the post-translational protein modifications affecting several catalytic and intracellular signaling pathways. Notably, the core pluripotency marker Oct4 is O-GlcNAcylated [11]. Subsequent studies have demonstrated that disruption of O-GlcNAcylation represses the transcriptional activity of Oct4 in mice [12] and humans [13]. Likewise, elevated O-GlcNAc levels caused by inhibition of O-GlcNAc hydrolase delay mouse embryonic stem cell (mESC) differentiation [14].
Glutamine and alpha-ketoglutarate
The TCA cycle is a central hub linking the metabolism of glucose, amino acids, and fatty acids. Metabolites derived from these metabolic pathways enter the TCA cycle, and TCA cycle metabolites are utilized for biosynthesis of other metabolites. One example of the utilization of TCA cycle metabolites is citrate, which is used for de novo fatty acid synthesis. In cancer cells, to refuel the TCA cycle, glutamine-derived α-ketoglutarate (αKG) enters the TCA cycle and replenishes oxaloacetate [15]. Furthermore, glutamine-derived malate is metabolized to pyruvate via malic enzyme 1 (NADP-dependent malic enzyme) to meet the high demand for NAPDH, which is also required as a cofactor for de novo fatty acid synthesis [15].
Similar to in cancer cells, glutamine plays key roles in hPSCs, which also exhibit increased de novo fatty acid synthesis. Interestingly, mitochondrial proteins aconitase 2 and isocitrate dehydrogenase 2/3, the enzymes that convert citrate to αKG, are downregulated in hPSCs. The physiological significance of downregulation of these enzymes is uncertain, but it may contribute to the activation of de novo fatty acid synthesis by damming citrate. Since aconitase 2 and isocitrate are downregulated in hPSCs, to sustain the metabolites of the latter part of the TCA cycle, that is, αKG to oxaloacetate, anaplerosis of the TCA cycle by glutamine-derived αKG is essential for the survival of hPSCs [16].
In addition to anaplerosis of the TCA cycle, glutamine is one of the substrates for glutathione (GSH) synthesis, and glutamine supports the stability of OCT4 in hPSCs by sustaining the production of GSH. In the absence of glutamine, endogenous GSH decreases, thereby increasing the oxidation of OCT4 cysteine residues, which results in OCT4 degradation [17].
Moreover, glutamine affects the epigenetic status of mESCs. A study of mESCs revealed that naïve mESCs rapidly consume exogenous glutamine and maintain a high αKG/succinate ratio to promote histone/DNA demethylation. αKG is a cofactor of JmjC domain-containing histone demethylation protein family (JHDM family) and ten-eleven translocation methylcytosine dioxygenase (TET). Both demethylation of trimethylated lysine-27 on histone H3 (H3K27me3) by the JHDM family and demethylation of DNA by TET are regarded as epigenetic modifications of active gene transcription [18].
These studies reveal the importance of glutamine and its metabolites in the survival and maintenance of pluripotency.
Tryptophan and kynurenine
Tryptophan, one of the essential aromatic amino acids, exhibits versatile roles in PSCs. Its metabolite, kynurenine, acts as a signaling molecule targeting the aryl hydrocarbon receptor (AHR), and two inconsistent studies in mESCs and hPSCs have been reported; mESCs maintain the AHR in a repressive state, and cells that failed to maintain their repressive state showed reduced levels of Oct4 and Sox2 [19]. However, in hPSCs, kynurenine stimulates binding of the AHR to putative promoter and enhancer regions of OCT4 and NANOG [20]. Studies on the roles of tryptophan and kynurenine are scarce and require further investigation.
S-adenosylmethionine, methionine, and one-carbon metabolism
mESCs catabolize threonine by threonine dehydrogenase and generate glycine and acetyl-CoA, which are utilized in one-carbon metabolism via the glycine cleavage system and TCA cycle, respectively [21]. Furthermore, these metabolites are utilized in the production of S-adenosylmethionine (SAM), which serves as a methyl donor in various biological reactions. Depletion of threonine from culture medium decreases SAM levels and leads to decreased trimethylation of histone H3 lysine 4 (H3K4me3), a methylation marker for the active state [22], resulting in increased differentiation.
A similar phenomenon can be observed in hPSCs. However, since threonine dehydrogenase is a pseudogene in humans, hPSCs cannot utilize threonine for the production of SAM; instead, hPSCs utilize methionine to maintain a high level of SAM. Transient deprivation of methionine decreases H3K4me3 and promotes cell differentiation, and long-term deprivation leads to p53-mediated cell death [23].
However, a study on naïve human embryonic stem cells (hESCs) showed that SAM needs to be controlled at low levels. In naïve hESCs, nicotinamide N-methyltransferase is upregulated, along with its product 1-methylnicotinamide. This enables a reduction of SAM levels and maintains a low level of H3K27me3, an epigenetic marker for repressive gene transcription and required for a naïve state [24]. Therefore, the role of SAM and its influence on epigenetic status in PSCs may be context dependent and must be interpreted with caution.
Lipids
De novo fatty acid synthesis is a process in which saturated fatty acid is synthesized from acetyl-CoA. Fatty acids are further elongated by elongase and/or desaturated by desaturases, and are utilized for the generation of glycerolipids (i.e., phosphatidylcholine, phosphatidylethanolamine, phosphatidylserine, and triacylglycerol) and sphingolipids (i.e., sphingomyelin and ceramide), which serve as the building blocks of phospholipid bilayers or energy storage. De novo fatty acid synthesis is upregulated in certain types of tumor cells to enable rapid cellular proliferation [25]. As for PSCs, during reprogramming of MEFs into miPSCs, levels of acetyl-CoA carboxylase and fatty acid synthase (FASN), the enzymes involved in de novo fatty acid synthesis, are upregulated. Pharmacological inhibition of these two enzymes results in decreased reprogramming efficiency [26]. Lipid synthesis, including de novo fatty acid synthesis and cholesterol synthesis, is regulated by sterol regulatory element-binding transcription factor 1 (Srebf1). Therefore, given that iPSCs activate lipid synthesis, one might expect that Srebf1 has a positive role in the reprogramming of somatic cells to iPSCs. Overexpression of Srebf1 reportedly enhanced reprogramming efficiency into miPSCs, while inhibition of Srebf1 reduced this efficiency. However, this regulatory effect does not arise from its regulation of lipid metabolism, but arises from its interaction with c-Myc, resulting in enhanced expression of pluripotent genes [27]. Interestingly, de novo fatty acid synthesis sustains mitochondrial dynamics in two ways in miPSCs, by producing fatty acids required for phospholipids, which are important components of the mitochondrial membrane, and by consuming fatty acid synthesis substrate acetyl-CoA. Consumption of acetyl-CoA prevents acetylation of mitochondrial fission 1 protein (FIS1) and acetylation-mediated ubiquitin-proteasome degradation of FIS1, leading to enhanced mitochondrial fission [28].
De novo fatty acid synthesis is not only crucial for reprogramming into miPSCs, but is also crucial for the survival of hPSCs. Knockdown of FASN, the final enzyme involved in de novo fatty acid synthesis, induces apoptosis in undifferentiated hPSCs. Inhibitors of FASN are similarly able to induce apoptosis in these cells. Interestingly, hiPSC-derived somatic cells, including cardiomyocytes (CMs), neurons, and hepatocytes, were not susceptible to FASN inhibition. These data suggest that FASN inhibition may be suitable for clinical regenerative medicine [29]. In undifferentiated hPSCs, oleate synthesis via SCD1 is upregulated; thus, its inhibition enables selective elimination of hPSCs [30].
Although mESCs and miPSCs share similar metabolic features, there are some differences. There is no notable difference in the metabolism of carbohydrates, hydroxy acid, free fatty acids, and the pentose phosphate pathway between mESCs and miPSCs. However, there are significant differences in phosphatidylcholine and phosphatidylethanolamine lipid structures, essential and non-essential amino acids, and polyamines [31].
Interestingly, hPSCs are able to modulate their glycolytic dependency under different culture conditions. hPSCs show a reduced dependency on glycolysis when cultured in the presence of feeder cells, and a greater dependency when cultured in feeder-free conditions [32]. This metabolic difference arises from the lipids contained in the culture media, since the lipids produced by feeder cells are consumed by the hPSCs and lipid supplementation is able to recapitulate the metabolic features of hPSCs grown with feeder cells in hPSCs without feeder cells. This data supports the idea that activated glycolysis provides biomass for cell proliferation. Another study reported a decreased dependency on glycolysis in primed hESCs cultured with MEFs [33]. Although this study did not specify the metabolite(s) needed to recapitulate the phenomenon, lipids are one of the candidate metabolites involved in this phenomenon. In addition, a recent report suggests that the lipid composition of the medium can alter the naïveness of hPSCs. Lipid deprivation in culture media skews hPSCs into a naïve-to-primed intermediate state [34]. Lipid deprivation-induced inhibition of extracellular signal-regulated kinase (ERK) signaling and skewed hPSCs toward the naïve-to-primed intermediate state.
Sphingolipids are a class of lipids containing sphingosine as a basal structure. De novo synthesis of sphingolipids starts from serine palmitoyl transferase, which consumes serine and palmitoyl-CoA as substrates. Ceramide is then glycosylated and diverges into various glycosphingolipids. The composition of glycosphingolipids varies between cell types, and hPSCs show a unique composition, with the glycosphingolipids stage-specific embryonic antigen (SSEA)-4 and SSEA-3 serving as PSC markers. Liang et al. reported the unique composition of glycosphingolipids, and that this composition changes during differentiation [35]. In hESCs, globo- and lacto-series glycosphingolipids are abundant. Along with embryoid body differentiation, these two types of glycosphingolipids decrease and are replaced with ganglio-series glycosphingolipids.
Mitochondria
Several studies have demonstrated that the mitochondria of PSCs are small and functionally immature compared to somatic cells [2, 36]. However, a study by Zhang et al. reported that hPSCs contain functional respiratory complexes and expression of uncoupling protein 2 (UCP2) in hPSCs decouple activated glycolysis and mitochondrial glucose oxidation [36]. Similarly, another group has reported that UCP2 exports TCA cycle metabolites, such as malate and oxaloacetate, from mitochondria [37].
Mitochondrial dynamics are crucial for the establishment of pluripotency and various mechanisms involved in the reprogramming of MEFs. In MEFs, OSKM (Oct4, Sox2, Klf4, and Myc) reprogramming causes Myc-induced cell proliferation, which leads to a decrease in mitochondria per cell, while OSK (Oct4, Sox2, and Klf4) reprogramming decreases mitochondria through another mechanism. In OSK reprogramming, mitochondrial mass initially increases and subsequently decreases through BNIP3L/NIX-mediated mitophagy [38].
Above all, it should be noted that it is the balance of mitochondrial dynamics (i.e., mitochondrial fission and fusion) that is important for pluripotency, and excess mitochondrial fission limits pluripotency. By balancing mitochondrial dynamics, Ca2+ homeostasis is maintained; therefore, CaMKII activity is balanced, leading to the accumulation of beta-catenin, which ultimately results in pluripotency in miPSCs [39].
Metabolism and differentiation
It is apparent that metabolism is not only passively altered during differentiation but also affects the cell fate of hPSCs. Here, we highlight the importance of metabolism in cell fate decision.
Glucose
In mESCs, culture conditions containing high glucose enhance cardiac differentiation through reactive oxygen species (ROS) production [40] (Fig. 2). Moreover, the addition of H2O2 is able to promote the differentiation of mESCs to CMs. Treatment with radical scavengers, such as trolox, pyrrolidine dithiocarbamate, and N-acetylcysteine, showed the opposite effect [41].

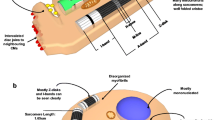
Metabolic regulation of hPSC cardiac differentiation and maturation. Undifferentiated hPSCs upregulate glycolysis, glutaminolysis, nucleotide synthesis, one-carbon metabolism, de novo fatty acid synthesis. Cardiac differentiation is enhanced by high glucose, ROS, fatty acylcarnitine, sphingosine-1-phosphate, and lysophosphatidic acid. On the other hand, glucose-derived acetyl-CoA inhibits differentiation. hPSC-CMs are metabolically immature and rely on lactate oxidation and glycolysis. By growing hPSC-CMs with fatty acids in the absence of glucose, hPSC-CMs metabolically mature and activate β oxidation and OXPHOS. Activation of HIF1α inhibits maturation. hPSC-CMs can be purified by utilizing the metabolic features of undifferentiated hPSCs and hPSC-CMs. Glucose- and glutamine-deprived, lactate-supplemented medium can purify hPSCs and eliminate undifferentiated hPSCs and non-cardiomyocytes. OXPHOS, oxidative phosphorylation
While the metabolic switch of PSCs from a glycolytic to an oxidative state during differentiation is well established, the specific timing of the transition is dependent on which lineage the cells are differentiating into. During differentiation to endoderm and mesoderm lineages, their metabolism switches to OXPHOS, but in the early phase of differentiation to ectoderm, high glycolytic flux is maintained, which requires MYC/MYCN in hPSCs [42].
Glycolysis-derived acetyl-CoA affects pluripotency and differentiation in mPSCs. Acetyl-CoA produced by glycolysis is used for histone acetylation, and inhibition of glycolysis decreases histone acetylation levels and promotes differentiation. Supplementation with acetate, which can be converted to acetyl-CoA, could delay differentiation [43].
Glutamine and αKG
While αKG maintains pluripotency through modification of histone and DNA demethylation, its ability to modify epigenetic status also affects differentiation. αKG promotes early differentiation in hPSCs, and reduced αKG delays differentiation. The accumulation of succinate, which is produced from αKG by histone or DNA demethylases, similarly, delays differentiation. Overall, the αKG/succinate ratio modifies histone methylation status [44].
However, the opposing effect of αKG on differentiation has been reported in a mESC study. There are three major pathways that generate αKG from glutamate, namely, glutamate dehydrogenase, alanine transaminase, and aspartate transaminase. However, phosphoserine aminotransferase 1 (Psat1), a serine synthesis pathway enzyme, also consumes glutamate and generates αKG. In mESCs, αKG produced by Psat1 alters H3K9me3 and H3K36me3 levels, and Psat1-knockdown accelerates differentiation [45].
These studies on the relationship between αKG and differentiation are controversial, but modulation of the serine synthesis pathway may alter one-carbon metabolism and, ultimately, histone and DNA methylation status through the production of SAM. Together with the difference in one-carbon metabolism, especially that of threonine, in mice and humans as described above, the somewhat opposing role of αKG on mouse and human PSCs needs further investigation.
Lipids
The lipid profile of mESCs shows abundant polyunsaturated fatty acids. Inhibition of polyunsaturated fatty acid metabolism, such as inhibition of Δ6/Δ-desaturase, phospholipase A2, cyclooxygenase 1/2, or lipoxygenase, results in delayed neuronal differentiation, which is defined by persistent expression of Oct4 and Nanog. Moreover, supplementation of the mitochondrial β-oxidation substrate acylcarnitine during differentiation promoted neuronal and cardiac differentiation [46]. This is consistent with reports that ROS promote cardiac differentiation, as mitochondrial β-oxidation produces ROS. In addition, sphingosine-1-phosphate and lysophosphatidic acid, both bioactive lipid species, are able to enhance cardiac differentiation in hiPSCs through nuclear accumulation of β-catenin, the canonical Wnt pathway mediator [47].
As for sphingolipids, during differentiation to the ectodermal lineage, globo- and lacto-series glycosphingolipids are replaced with ganglioside-series glycosphingolipids. During differentiation to the endodermal lineage, Gb4 ceramide dominates [48]. It has been reported that the abundance of globo-series glycosphingolipids accounts for the expression of neuronal transcription factors. Globo-series sphingolipids reside in the promoter region of autism susceptibility candidate 2 (AUTS2), and along with neuronal differentiation, reduction in globo-series glycosphingolipids leads to the induction of AUTS2, and AUTS2 activates the production of ganglio-series glycosphingolipids and other neuronal genes [49].
Exploiting the metabolic signature of hPSCs in regenerative medicine
Regenerative medicine using hPSCs is an attractive therapy for diseases resistant to conventional medical therapies. However, several limitations must be overcome to achieve full clinical application. Among them, tumor formation by residual undifferentiated cells is of great importance, and while several strategies have been proposed to overcome this limitation, many are not ideal for clinical application due to high cost and poor scalability. Our research group has developed a strategy based on the distinct metabolic features of hPSCs; the different metabolic features of undifferentiated hPSCs and hPSC-derived CMs (hPSC-CMs), and their potential application, will here-after be discussed (Fig. 2).
hPSC-CMs
hPSC-CMs are immature and resemble that of the fetus in terms of their size, sarcomere structure, contractile activity, electrophysiological activity, mitochondrial structure, and metabolism [50]. Mature CMs rely mainly on fatty acid oxidation for energy production, while fetal CMs rely mainly on glycolysis and lactate oxidation [51]. Although the metabolism of hPSC-CMs is similar to that of fetal CMs and immature compared to adult CMs, differentiation of hPSCs to hPSC-CMs is accompanied by a transition from aerobic glycolysis to mitochondrial oxidation, and it greatly differs from that of hPSCs and hPSC-derived non-CMs, which are generated during cardiac differentiation [52]. Therefore, the difference in metabolism can be exploited to purify hPSC-CMs after differentiation from hPSCs. Undifferentiated hPSCs and hPSC-derived non-CMs rely largely on glucose and glutamine metabolism, and their deprivation leads to cell death. hPSC-CMs are able to survive under glucose and glutamine deprivation conditions, in the presence of lactate, whereby hPSC-CMs consume lactate to fuel the TCA cycle and produce energy. Therefore, in glucose- and glutamine-deprived, lactate-supplemented culture medium, undifferentiated hPSCs and hPSC-derived non-CMs cannot survive, whereas hPSC-CMs can survive, thus allowing for their purification. This method is particularly promising in cardiac regeneration therapy using hPSC-CMs because it has advantages in cost, ease-of-use, and scalability [16, 53, 54].
As described previously, the mitochondria of undifferentiated hPSCs are small and immature. Mitochondrial metabolism (i.e., the TCA cycle, fatty acid oxidation, and OXPHOS) in hPSC-CMs is immature compared to adult CMs, but hPSC-CMs contain larger amounts of mitochondria than undifferentiated hPSCs. Moreover, mitochondria of hPSC-CMs are larger in size and more mature than those of undifferentiated hPSCs. Therefore, fluorescence-assisted cell sorting of hPSC-CMs by tetramethylrhodamine methyl ester perchlorate (TMRM) staining enables efficient purification of hPSC-CMs [55].
The immaturity of hPSC-CMs may limit their application in regenerative therapy, drug discovery, and disease modeling. Long-term culture of hPSC-CMs improves maturation, but it has been reported that hPSC-CMs after long-term culture cannot reach the level of maturation of adult CMs [56, 57]. Therefore, to facilitate maturation, several methods have been reported, including culture substrates, mechanical stress, electrical stimulation, and thyroid hormones [58,59,60,61]. Of these, metabolism is one of the more promising strategies. Metabolism is an important factor influencing cell properties and can be further exploited in the maturation of hPSC-CMs. Fatty acid oxidation is a hallmark metabolic feature of adult CMs, and glucose-deprived and fatty acids (palmitic acid and oleic acid)-supplemented culture medium can facilitate maturation [62, 63]. However, excess saturated fatty acids (i.e., palmitic acid) may cause lipotoxicity [64]. Galactose supplementation to glucose-deprived and fatty acid-supplemented medium ameliorated lipotoxicity caused by saturated fatty acids and enhanced maturation [65]. Cultures with glucose-containing medium upregulate the HIF1α-lactate dehydrogenase A axis and leads to active glycolysis. By inhibiting HIF1α or lactate dehydrogenase A with small molecules, structural, metabolic, and electrophysiological maturation was achieved [65]. Another study reported that high-glucose culture conditions inhibit the maturation of hPSC-CMs via the pentose phosphate pathway [66]. Overall, these findings underscore the importance of metabolism and the need for optimization of culture conditions to facilitate the maturation of hPSC-CMs.
Conclusions
In this study, we have detailed several studies reporting on the distinct metabolic features of hPSCs and how metabolism can affect the properties of hPSCs and their derivatives. While the clinical application of regenerative medicine using hPSC-derived somatic cells is gaining momentum, various cost and safety concerns remain to be addressed. Exploiting the metabolic characteristics governing cells at different stages may serve as an effective strategy to mitigate these issues.
Availability of data and materials
Not applicable.
Abbreviations
- αKG:
-
α-ketoglutarate
- AHR:
-
Aryl hydrocarbon receptor
- AUTS2:
-
Autism susceptibility candidate 2
- ERRα/γ:
-
Estrogen-related receptor alpha/gamma
- ERK:
-
Extracellular signal-regulated kinase
- ESC:
-
Embryonic stem cell
- FASN:
-
Fatty acid synthase
- GSH:
-
Glutathione
- HBP:
-
Hexosamine biosynthetic pathway
- hESC:
-
Human embryonic stem cell
- HIF:
-
Hypoxia-inducible factor
- hiPSC:
-
Human induced pluripotent stem cells
- hPSC:
-
Human pluripotent stem cell
- iPSC:
-
Induced pluripotent stem cell
- JHDM family:
-
JmjC domain-containing histone demethylation protein family
- mESC:
-
Mouse embryonic stem cell
- miPSC:
-
Mouse induced pluripotent stem cell
- mPSC:
-
Mouse pluripotent stem cell
- NAD+ :
-
Nicotinamide adenine dinucleotide
- OXPHOS:
-
Oxidative phosphorylation
- PGC-1α/β:
-
Peroxisome proliferator-activated receptor gamma coactivator 1-alpha/beta
- PSC:
-
Pluripotent stem cell
- Srebf1:
-
Sterol regulatory element binding transcription factor 1
- SSEA:
-
Stage specific embryonic antigen
- TCA cycle:
-
Tricarboxylic acid cycle
- TET:
-
Ten-eleven translocation methylcytosine dioxygenase
- UDP-GlcNAc:
-
Uridine diphosphate N-acetylglucosamine
References
Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–14.
Kondoh H, Lleonart ME, Nakashima Y, Yokode M, Tanaka M, Bernard D, et al. A high glycolytic flux supports the proliferative potential of murine embryonic stem cells. Antioxid Redox Signal. 2007;12(587):eaaw3306.
Varum S, Rodrigues AS, Moura MB, Momcilovic O, Easley CA, Ramalho-Santos J, et al. Energy metabolism in human pluripotent stem cells and their differentiated counterparts. Plos One. 2011;6(6):e20914.
Panopoulos AD, Yanes O, Ruiz S, Kida YS, Diep D, Tautenhahn R, et al. The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell Res. 2012;22(1):168–77.
Kim H, Jang H, Kim TW, Kang BH, Lee SE, Jeon YK, et al. Core pluripotency factors directly regulate metabolism in embryonic stem cell to maintain pluripotency. Stem Cells. 2015;33(9):2699–711.
Lee J, Kim HK, Han YM, Kim J. Pyruvate kinase isozyme type M2 (PKM2) interacts and cooperates with Oct-4 in regulating transcription. Int J Biochem Cell Biol. 2008;40(5):1043–54.
Folmes CD, Nelson TJ, Martinez-Fernandez A, Arrell DK, Lindor JZ, Dzeja PP, et al. Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab. 2011;14(2):264–71.
Mathieu J, Zhou W, Xing Y, Sperber H, Ferreccio A, Agoston Z, et al. Hypoxia-inducible factors have distinct and stage-specific roles during reprogramming of human cells to pluripotency. Cell Stem Cell. 2014;14(5):592–605.
Prigione A, Rohwer N, Hoffmann S, Mlody B, Drews K, Bukowiecki R, et al. HIF1alpha modulates cell fate reprogramming through early glycolytic shift and upregulation of PDK1-3 and PKM2. Stem Cells. 2014;32(2):364–76.
Kida YS, Kawamura T, Wei Z, Sogo T, Jacinto S, Shigeno A, et al. ERRs mediate a metabolic switch required for somatic cell reprogramming to pluripotency. Cell Stem Cell. 2015;16(5):547–55.
Webster DM, Teo CF, Sun Y, Wloga D, Gay S, Klonowski KD, et al. O-GlcNAc modifications regulate cell survival and epiboly during zebrafish development. BMC Dev Biol. 2009;9:28.
Jang H, Kim TW, Yoon S, Choi SY, Kang TW, Kim SY, et al. O-GlcNAc regulates pluripotency and reprogramming by directly acting on core components of the pluripotency network. Cell Stem Cell. 2012;11(1):62–74.
Constable S, Lim JM, Vaidyanathan K, Wells L. O-GlcNAc transferase regulates transcriptional activity of human Oct4. Glycobiology. 2017;27(10):927–37.
Speakman CM, Domke TC, Wongpaiboonwattana W, Sanders K, Mudaliar M, van Aalten DM, et al. Elevated O-GlcNAc levels activate epigenetically repressed genes and delay mouse ESC differentiation without affecting naive to primed cell transition. Stem Cells. 2014;32(10):2605–15.
DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA. 2007;104(49):19345–50.
Tohyama S, Fujita J, Hishiki T, Matsuura T, Hattori F, Ohno R, et al. Glutamine oxidation is indispensable for survival of human pluripotent stem cells. Cell Metab. 2016;23(4):663–74.
Marsboom G, Zhang GF, Pohl-Avila N, Zhang Y, Yuan Y, Kang H, et al. Glutamine metabolism regulates the pluripotency transcription factor OCT4. Cell Rep. 2016;16(2):323–32.
Carey BW, Finley LW, Cross JR, Allis CD, Thompson CB. Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature. 2015;518(7539):413–6.
Ko CI, Fan Y, de Gannes M, Wang Q, Xia Y, Puga A. Repression of the aryl hydrocarbon receptor is required to maintain mitotic progression and prevent loss of pluripotency of embryonic stem cells. Stem Cells. 2016;34(12):2825–39.
Yamamoto T, Hatabayashi K, Arita M, Yajima N, Takenaka C, Suzuki T, et al. Kynurenine signaling through the aryl hydrocarbon receptor maintains the undifferentiated state of human embryonic stem cells. Sci Signal. 2019;12(587):eaaw3306.
Wang J, Alexander P, Wu L, Hammer R, Cleaver O, McKnight SL. Dependence of mouse embryonic stem cells on threonine catabolism. Science. 2009;325(5939):435–9.
Shyh-Chang N, Locasale JW, Lyssiotis CA, Zheng Y, Teo RY, Ratanasirintrawoot S, et al. Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science. 2013;339(6116):222–6.
Shiraki N, Shiraki Y, Tsuyama T, Obata F, Miura M, Nagae G, et al. Methionine metabolism regulates maintenance and differentiation of human pluripotent stem cells. Cell Metab. 2014;19(5):780–94.
Sperber H, Mathieu J, Wang Y, Ferreccio A, Hesson J, Xu Z, et al. The metabolome regulates the epigenetic landscape during naive-to-primed human embryonic stem cell transition. Nat Cell Biol. 2015;17(12):1523–35.
Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7(10):763–77.
Vazquez-Martin A, Corominas-Faja B, Cufi S, Vellon L, Oliveras-Ferraros C, Menendez OJ, et al. The mitochondrial H(+)-ATP synthase and the lipogenic switch: new core components of metabolic reprogramming in induced pluripotent stem (iPS) cells. Cell Cycle. 2013;12(2):207–18.
Wu Y, Chen K, Liu X, Huang L, Zhao D, Li L, et al. Srebp-1 interacts with c-Myc to enhance somatic cell reprogramming. Stem Cells. 2016;34(1):83–92.
Wang L, Zhang T, Wang L, Cai Y, Zhong X, He X, et al. Fatty acid synthesis is critical for stem cell pluripotency via promoting mitochondrial fission. EMBO J. 2017;36(10):1330–47.
Tanosaki S, Tohyama S, Fujita J, Someya S, Hishiki T, Matsuura T, et al. Fatty acid synthesis is indispensable for survival of human pluripotent stem cells. iScience. 2020;23(9):101535.
Ben-David U, Gan QF, Golan-Lev T, Arora P, Yanuka O, Oren YS, et al. Selective elimination of human pluripotent stem cells by an oleate synthesis inhibitor discovered in a high-throughput screen. Cell Stem Cell. 2013;12(2):167–79.
Meissen JK, Yuen BT, Kind T, Riggs JW, Barupal DK, Knoepfler PS, et al. Induced pluripotent stem cells show metabolomic differences to embryonic stem cells in polyunsaturated phosphatidylcholines and primary metabolism. Plos One. 2012;7(10):e46770.
Zhang H, Badur MG, Divakaruni AS, Parker SJ, Jager C, Hiller K, et al. Distinct metabolic states can support self-renewal and lipogenesis in human pluripotent stem cells under different culture conditions. Cell Rep. 2016;16(6):1536–47.
Gu W, Gaeta X, Sahakyan A, Chan AB, Hong CS, Kim R, et al. Glycolytic metabolism plays a functional role in regulating human pluripotent stem cell state. Cell Stem Cell. 2016;19(4):476–90.
Cornacchia D, Zhang C, Zimmer B, Chung SY, Fan Y, Soliman MA, et al. Lipid deprivation induces a stable, naive-to-primed intermediate state of pluripotency in human PSCs. Cell Stem Cell. 2019;25(1):120–36 e10.
Liang YJ, Kuo HH, Lin CH, Chen YY, Yang BC, Cheng YY, et al. Switching of the core structures of glycosphingolipids from globo- and lacto- to ganglio-series upon human embryonic stem cell differentiation. Proc Natl Acad Sci USA. 2010;107(52):22564–9.
Zhang J, Khvorostov I, Hong JS, Oktay Y, Vergnes L, Nuebel E, et al. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 2011;30(24):4860–73.
Vozza A, Parisi G, De Leonardis F, Lasorsa FM, Castegna A, Amorese D, et al. UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc Natl Acad Sci USA. 2014;111(3):960–5.
Xiang G, Yang L, Long Q, Chen K, Tang H, Wu Y, et al. BNIP3L-dependent mitophagy accounts for mitochondrial clearance during 3 factors-induced somatic cell reprogramming. Autophagy. 2017;13(9):1543–55.
Zhong X, Cui P, Cai Y, Wang L, He X, Long P, et al. Mitochondrial dynamics is critical for the full pluripotency and embryonic developmental potential of pluripotent stem cells. Cell Metab. 2019;29(4):979–92 e4.
Crespo FL, Sobrado VR, Gomez L, Cervera AM, McCreath KJ. Mitochondrial reactive oxygen species mediate cardiomyocyte formation from embryonic stem cells in high glucose. Stem Cells. 2010;28(7):1132–42.
Sauer H, Rahimi G, Hescheler J, Wartenberg M. Role of reactive oxygen species and phosphatidylinositol 3-kinase in cardiomyocyte differentiation of embryonic stem cells. FEBS Lett. 2000;476(3):218–23.
Cliff TS, Wu T, Boward BR, Yin A, Yin H, Glushka JN, et al. MYC Controls human pluripotent stem cell fate decisions through regulation of metabolic flux. Cell Stem Cell. 2017;21(4):502–16 e9.
Moussaieff A, Rouleau M, Kitsberg D, Cohen M, Levy G, Barasch D, et al. Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell Metab. 2015;21(3):392–402.
TeSlaa T, Chaikovsky AC, Lipchina I, Escobar SL, Hochedlinger K, Huang J, et al. alpha-Ketoglutarate accelerates the initial differentiation of primed human pluripotent stem cells. Cell Metab. 2016;24(3):485–93.
Hwang IY, Kwak S, Lee S, Kim H, Lee SE, Kim JH, et al. Psat1-Dependent fluctuations in alpha-ketoglutarate affect the timing of ESC differentiation. Cell Metab. 2016;24(3):494–501.
Yanes O, Clark J, Wong DM, Patti GJ, Sanchez-Ruiz A, Benton HP, et al. Metabolic oxidation regulates embryonic stem cell differentiation. Nat Chem Biol. 2010;6(6):411–7.
Sharma A, Zhang Y, Buikema JW, Serpooshan V, Chirikian O, Kosaric N, et al. Stage-specific effects of bioactive lipids on human iPSC cardiac differentiation and cardiomyocyte proliferation. Sci Rep. 2018;8(1):6618.
Liang YJ, Yang BC, Chen JM, Lin YH, Huang CL, Cheng YY, et al. Changes in glycosphingolipid composition during differentiation of human embryonic stem cells to ectodermal or endodermal lineages. Stem Cells. 2011;29(12):1995–2004.
Russo D, Della Ragione F, Rizzo R, Sugiyama E, Scalabri F, Hori K, et al. Glycosphingolipid metabolic reprogramming drives neural differentiation. EMBO J. 2018;37(7):e97674.
Marchiano S, Bertero A, Murry CE. Learn from your elders: developmental biology lessons to guide maturation of stem cell-derived cardiomyocytes. Pediatr Cardiol. 2019;40(7):1367–87.
Lopaschuk GD, Jaswal JS. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J Cardiovasc Pharmacol. 2010;56(2):130–40.
Chung S, Dzeja PP, Faustino RS, Perez-Terzic C, Behfar A, Terzic A. Mitochondrial oxidative metabolism is required for the cardiac differentiation of stem cells. Nat Clin Pract Cardiovasc Med. 2007;4(Suppl 1):S60–7.
Tohyama S, Hattori F, Sano M, Hishiki T, Nagahata Y, Matsuura T, et al. Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell. 2013;12(1):127–37.
Tohyama S, Fujita J, Fujita C, Yamaguchi M, Kanaami S, Ohno R, et al. Efficient large-scale 2D culture system for human induced pluripotent stem cells and differentiated cardiomyocytes. Stem Cell Reports. 2017;9(5):1406–14.
Hattori F, Chen H, Yamashita H, Tohyama S, Satoh YS, Yuasa S, et al. Nongenetic method for purifying stem cell-derived cardiomyocytes. Nat Methods. 2010;7(1):61–6.
Kamakura T, Makiyama T, Sasaki K, Yoshida Y, Wuriyanghai Y, Chen J, et al. Ultrastructural maturation of human-induced pluripotent stem cell-derived cardiomyocytes in a long-term culture. Circ J. 2013;77(5):1307–14.
Lundy SD, Zhu WZ, Regnier M, Laflamme MA. Structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Stem Cells Dev. 2013;22(14):1991–2002.
Feaster TK, Cadar AG, Wang L, Williams CH, Chun YW, Hempel JE, et al. Matrigel mattress: a method for the generation of single contracting human-induced pluripotent stem cell-derived cardiomyocytes. Circ Res. 2015;117(12):995–1000.
Ruan JL, Tulloch NL, Saiget M, Paige SL, Razumova MV, Regnier M, et al. Mechanical stress promotes maturation of human myocardium from pluripotent stem cell-derived progenitors. Stem Cells. 2015;33(7):2148–57.
Nunes SS, Miklas JW, Liu J, Aschar-Sobbi R, Xiao Y, Zhang B, et al. Biowire: a platform for maturation of human pluripotent stem cell-derived cardiomyocytes. Nat Methods. 2013;10(8):781–7.
Yang X, Rodriguez M, Pabon L, Fischer KA, Reinecke H, Regnier M, et al. Tri-iodo-l-thyronine promotes the maturation of human cardiomyocytes-derived from induced pluripotent stem cells. J Mol Cell Cardiol. 2014;72:296–304.
Yang X, Rodriguez ML, Leonard A, Sun L, Fischer KA, Wang Y, et al. Fatty acids enhance the maturation of cardiomyocytes derived from human pluripotent stem cells. Stem Cell Rep. 2019;13(4):657–68.
Ramachandra CJA, Mehta A, Wong P, Ja K, Fritsche-Danielson R, Bhat RV, et al. Fatty acid metabolism driven mitochondrial bioenergetics promotes advanced developmental phenotypes in human induced pluripotent stem cell derived cardiomyocytes. Int J Cardiol. 2018;272:288–97.
Yamamoto T, Endo J, Kataoka M, Matsuhashi T, Katsumata Y, Shirakawa K, et al. Palmitate induces cardiomyocyte death via inositol requiring enzyme-1 (IRE1)-mediated signaling independent of X-box binding protein 1 (XBP1). Biochem Biophys Res Commun. 2020;526(1):122–7.
Correia C, Koshkin A, Duarte P, Hu D, Teixeira A, Domian I, et al. Distinct carbon sources affect structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Sci Rep. 2017;7(1):8590.
Nakano H, Minami I, Braas D, Pappoe H, Wu X, Sagadevan A, et al. Glucose inhibits cardiac muscle maturation through nucleotide biosynthesis. eLIFE. 2017;6:e29330.
Acknowledgements
Not applicable.
Funding
The present work was mainly supported by Projects for Technological Development, the Research Center Network for Realization of Regenerative Medicine by Japan, and the Japan Agency for Medical Research and Development (grant number: 20bm0404023h0003, to S. Tohyama).
Author information
Authors and Affiliations
Contributions
S. Tanosaki wrote the original manuscript. S. Tohyama reviewed and edited the manuscript. S. Tohyama and K.F. acquired the funding and supervised the study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
S. Tohyama, J.F., and K.F. own equity in Heartseed, Inc. K.F. is the CEO of Heartseed, Inc. S. Tanosaki has no conflicts of interest to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tanosaki, S., Tohyama, S., Kishino, Y. et al. Metabolism of human pluripotent stem cells and differentiated cells for regenerative therapy: a focus on cardiomyocytes. Inflamm Regener 41, 5 (2021). https://doi.org/10.1186/s41232-021-00156-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41232-021-00156-9