
Abstract
Hematopoietic stem cells (HSCs) have self-renewal capacity and differentiation potential into all lineages of blood cells throughout the lifetime of an organism. The function of HSCs gradually changes during aging. To date, various stress factors influencing HSC aging have been identified. The increased production of reactive oxygen species and DNA damage responses are causatively attributed to HSC aging. The increased apolarity is a prominent feature of aged HSCs, whereas it is less obvious in young HSCs. The bone marrow (BM) microenvironment niche is a crucial factor for HSC aging. Mesenchymal stem cells show skewed differentiation during aging, which leads to decreased bone formation and increased adipogenesis. The accumulation of adipocytes confers negative effects on hematopoiesis. Loss of sympathetic nerve fibers or adrenoreceptor β3 signaling induces premature HSC and niche aging. Epigenetic regulators such as polycomb group proteins and the sirtuin family of proteins act to prevent premature aging. Targeting these factors, several rejuvenation strategies for aged HSCs have been employed in mice. However, we still do not know whether these strategies can be extrapolated to human HSCs. Aging is frequently accompanied by the development of clonal hematopoiesis, which is called age-related clonal hematopoiesis (ARCH) or clonal hematopoiesis of indeterminate potential (CHIP). Most ARCH/CHIP mutations occur in genes encoding epigenetic regulators including DNMT3A, TET2, and ASXL1, which suggests the relevance of epigenetic drift during the aging process. ARCH/CHIP is a strong risk factor for subsequent hematologic cancer. Notably, it also has an impact on the development of non-malignant disorders such as coronary heart disease. Further studies are warranted to decipher the complete picture of molecular crosstalk that regulates HSC aging.
Similar content being viewed by others

Background
Hematopoietic stem cells (HSCs) are characterized by their ability to self-renew and their potential to differentiate into all lineages of blood cells throughout the lifetime of an organism. During aging, the functionality of HSCs gradually changes in mice as well as humans. Aged HSCs show several characteristic alterations in phenotype, such as an increase in the frequency of immunophenotypic HSCs, a decrease in regenerative capacity [1,2,3], myeloid-biased differentiation [1, 2], impaired homing and engraftment upon transplantation [4], platelet-biased differentiation [5], and megakaryocytic/erythroid-biased gene expression patterns [6]. Additionally, in humans, age-associated hematopoietic changes are correlated with an increased incidence of myeloid malignancies [7, 8]. In this review, we summarize the current knowledge on the mechanisms of HSC aging and clonal hematopoiesis, as well as the results of recent single-cell profiling studies on aged HSCs.
Main text
Factors contributing to HSC aging
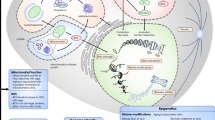
Over the past decade, the multiple differences between young and aged HSCs have been examined, and various stress factors influencing HSC aging have been identified [8,9,10,11,12,13]. Functional decline in aged HSCs is associated with changes in cell-intrinsic aging drivers, such as metabolic alternations, impaired autophagy, proteostasis, DNA damage, senescence, and other signaling pathways (mTOR, WNT, Janus kinase, and signal transducer and activator of transcription, NF-κB, and TGF-β). Loss of epigenetic regulation (DNA methylation, histone marks, and chromatin architecture) and cell polarity are additional cell-intrinsic features. Additionally, cell-extrinsic factors, including the development of a proinflammatory milieu and decreased function of the old bone marrow (BM) niche, contribute to HSC aging. Here, we focus on the current understanding of these factors, particularly reactive oxygen species (ROS), DNA damage, polarity, stem cell niches, senescence, and epigenetics (Fig. 1).

Factors that promote HSC aging and rejuvenation strategies for aged HSCs. N-acetyl cysteine (NAC) and p38 mitogen-activated protein kinase (MAPK) inhibitor rejuvenate aged hematopoietic stem cells (HSCs) by reducing reactive oxygen species (ROS) levels. Cdc42 activity inhibitor (CASIN) increases the percentage of polarized cells, restores the spatial distribution of H4 at lysine 16 acetylation, increases lymphoid output, and reduces myeloid lineage output. Sirt3 overexpression restores the long-term competitive repopulation ability. Sirt7 overexpression rescues myeloid-biased differentiation. ABT263 (navitoclax), a BCL-2- and BCL-xL-specific inhibitor, selectively kills senescent cells. Supplementation of a sympathomimetic acting selectively on adrenoreceptor β3 rejuvenates the function of aged HSCs. The red and blue arrows indicate aging and rejuvenation, respectively
ROS
The maintenance of appropriate ROS levels is critical for the maintenance of HSCs and DNA integrity, as well as for preventing ROS-mediated cell damage [14]. Jang and Sharkis reported that HSCs with low levels of ROS had high potential for self-renewal, which was attenuated during aging as the ROS levels increased [15]. A ROS inhibitor—N-acetyl cysteine (NAC)—and a p38 mitogen-activated protein kinase (MAPK) inhibitor have been shown to rescue the decreased functionality of HSCs with high levels of ROS, demonstrating a role of ROS and p38 MAPK in ROS-mediated HSC maintenance.
Forkhead O (FOXO) family transcription factors, which are important downstream targets of the PI3K-AKT pathway, play a significant role in mediating the response to physiological oxidative stress and are essential for the self-renewal of HSCs. The HSCs of mice deficient in Foxo1, Foxo3a, and Foxo4a show a marked increase in ROS levels due to the dysregulation of genes that regulate ROS [16]. Miyamoto et al. generated gene-targeted Foxo3a−/− mice and showed that HSC frequencies were significantly decreased in aged Foxo3a−/− mice than in the littermate controls [17].
DNA damage
DNA damage is caused by various factors, such as radiation, exogenous genotoxins, and free radicals, and has been observed to accumulate during aging in many studies [18,19,20]. Phosphorylation of histone H2AX (γ-H2AX) is an indicator of DNA damage, and HSCs accumulate γ-H2AX signals with age [21]. Older HSCs which are still undergoing the cell cycle show heightened levels of replication-associated γ-H2AX foci and increased levels of replication stress associated with cell cycle defects, altered DNA fork replication dynamics, and chromosome gaps or breaks [22]. Nonetheless, once old HSCs re-establish quiescence, residual replication stress on ribosomal DNA genes leads to the formation of nucleolar-associated γH2AX signals, which persist owing to ineffective H2AX dephosphorylation rather than ongoing DNA damage. Thus, persistent nucleolar γH2AX acts as a histone modification, marking the transcriptional silencing of rDNA genes and decreased ribosome biogenesis in quiescent old HSCs.
The DNA damage response is mainly regulated by the following pathways: base excision repair, nucleotide excision repair (NER), homologous recombination repair (HR), and non-homologous end joining repair (NHEJ) [21,22,23,24,25]. The NER pathway maintains HSC functionality during aging by preserving the cells’ ability for reconstitution, self-renewal, and proliferation, and by preventing programmed cell death under conditions of stress [21]. The NER pathway-associated gene Xab2 has been reported to be downregulated in aged HSCs, suggesting that the NER pathway acts to rescue HSC function, but its activity decreases during aging [23]. Further analysis revealed that quiescent hematopoietic stem and progenitor cells (HSPCs) undergo error-prone NHEJ-mediated DNA repair while proliferating HSPCs do HR-mediated DNA repair [24]. HR-mediated DNA repair uses a template for accurate repair, typically the sister chromatid, and thus can only occur in cycling cells. In contrast, NHEJ-mediated DNA repair has a limited requirement for sequence homology and can occur during any stage of the cell cycle [25]. The accumulation of NHEJ-mediated mutations impairs HSC performance and is a major factor in the loss of function observed in aged HSCs.
Polarity and WNT signaling
A reduction in the number of cells with a polar distribution of microtubules has been observed in aged early hematopoietic progenitor cells [26]. Florian et al. reported that elevated activity of the small Rho GTPase Cdc42 is correlated with the loss of polarity in aged HSCs [27]. A Cdc42 activity inhibitor (CASIN) rejuvenated the functionality of aged HSCs, improved the percentage of polarized cells in aged HSCs, and restored the level and spatial distribution of histone H4 at lysine 16 acetylation (H4K16ac), which has been found to be reduced in multiple tissues in aged mice.
The WNT pathway proteins function as proliferation-inducing growth factors and affect cell-fate decisions, apoptosis, and quiescence [28]. WNT3a—which is associated with the canonical pathway—and WNT5a—which is associated with the non-canonical pathway—are the most widely studied proteins in hematopoiesis. The former promotes lymphopoiesis, and the latter enhances myelopoiesis [29]. Florian et al. reported that elevated expression of Wnt5a in aged HSCs results in apolarity through increased Cdc42 activity [30]. The activation of Cdc42 by Wnt5a treatment-induced aging-related stem cell apolarity, reduction of the self-renewal potential, and myeloid-skewed differentiation in young HSCs. Povinelli and Nemeth reported that inhibition of the Wnt ligand receptor Ryk blocked the ability of WNT5a to induce HSC quiescence, and enhanced short- and long-term hematopoietic repopulation through reduction of the intracellular levels of ROS [31].
Stem cell niches
Niche factors secreted from aging stromal cells play a critical role in shifting the subtype of HSCs. RANTES (CCL5) is a key proinflammatory cytokine that accumulates in aged BM and is expressed by stromal as well as differentiated blood cells [32]. Ergen et al. reported that a brief ex vivo exposure of HSCs to Rantes resulted in T cell deficiency and expanded the myeloid progenitors. In contrast, the deletion of Rantes in mice rescued myeloid-biased differentiation and increased T cell production [33].
An additional hallmark of aging is altered HSC distribution. Aging induces significant remodeling of HSC niches. Mesenchymal stem cells exhibit skewed differentiation during aging, leading to decreased bone formation and increased adipogenesis [34, 35]. The accumulation of adipocytes in the BM negatively affects hematopoiesis [36,37,38]. Arterioles and transitional (type H) vessels that connect to the arterioles are found in direct proximity and share many features that distinguish them from BM sinusoidal (type L) capillaries. Type H capillaries are surrounded by osteoprogenitors, and both cells as well as arterioles decline significantly with aging [39, 40]. Activation of endothelial Notch signaling can restore the numbers of arteries and type H capillaries, although HSC function is not rejuvenated [40]. Aged mice also show a significant increase in vascular leakiness and ROS levels in endothelial cells in BM [41].
The sympathetic nervous system (SNS) regulates the attraction of stem cells to the microenvironment by orchestrating the release of adrenergic neurotransmitter in a circadian manner [42, 43]. Maryanovich et al. reported that loss of sympathetic nerves around the arteriolar niche, or adrenoreceptor β3 signaling, induced the premature aging of HSCs and niche, indicating that BM innervation by the SNS regulates the aging of HSCs and their niche [44]. Supplementation of a sympathomimetic acting selectively on the adrenoreceptor β3 of aged mice significantly rejuvenated the function of aged HSCs.
Senescence
Senescent cells show a characteristic phenotype known as the senescence-associated secretory phenotype (SASP), in which the cells secrete high levels of proinflammatory and matrix-degrading molecules [45]. Two major pathways are implicated in the induction of senescence, tumor suppressors p16Ink4a and p19Arf (p14ARF in humans) that are encoded by the genes at the Ink4a/Arf (Cdkn2a) locus [46]. p19Arf stabilizes p53 by inhibiting MDM2, resulting in the activation of p53 target genes involved in cell cycle arrest and apoptosis. p16Ink4a blocks the assembly of catalytically active cyclin D-CDK4/6 complexes by maintaining Rb in a hypophosphorylated state. p16Ink4a expression in HSCs increases with age, and the absence of p16Ink4a could mitigate the repopulating defects in HSCs [47]. Chang et al. reported that ABT263 (navitoclax), a BCL-2- and BCL-xL-specific inhibitor, selectively induces the apoptosis of senescent HSCs after total body irradiation (TBI), mitigating TBI-induced premature aging of the hematopoietic system, and rejuvenating aged HSCs in normally aged mice [48].
Epigenetics
Stem cells have developed epigenetic regulatory mechanisms to prevent premature aging [49]. Polycomb group (PcG) proteins are representative of such regulators. The PcG protein complexes repress the expression of their target genes and are divided into the polycomb repressive complex 1 (PRC1) and PRC2. The former mono-ubiquitinates histone H2A at lysine 119 (H2AK119Ub), and the latter trimethylates histone H3 at lysine 27 (H3K27me3) [50]. PcG complexes target the Cdkn2a locus and regulate HSCs by acting as critical failsafe against the premature aging induced by the p16Ink4a and p19Arf tumor suppressors [51,52,53,54,55]. Forced expression of PcG genes, such as Ezh2, Kdm2b/Fbxl10, Bmi1, and Cbx7, promotes HSC self-renewal and preserves HSC potential during serial transplantations, an observation which is also indicative of their role in HSC aging [56,57,58,59,60]. It remains to be investigated how changes in these genes contribute to HSC aging other than by their impact on the Ink4a/Arf locus.
Members of the sirtuin family of proteins are NAD+-dependent deacetylases, which play key roles in responding to nutritional and environmental perturbations such as DNA damage and oxidative stress [61]. Mammals have seven sirtuins, SIRT1–7: SIRT1, SIRT6, and SIRT7 in the nucleus; SIRT3, SIRT4, and SIRT5 in the mitochondria; and SIRT2 in the cytoplasm [62]. In HSPCs, Sirt1 ablation increases Hoxa9 expression and HSPC expansion, depending on hematopoietic stress [63]. However, it causes genomic instability as well as the accumulation of DNA damage and eventually results in a loss of long-term HSPCs.
Sirt3 deacetylates and activates mitochondrial isocitrate dehydrogenase 2, leading to increased levels of NADPH and an increased ratio of reduced-to-oxidized glutathione in the mitochondria [64]. The levels of Sirt3 are reduced during aging, and its overexpression rescues functional defects in aged HSCs in mice [65]. Sirt7 expression is also reduced in aged HSCs, while its overexpression increases the reconstitution capacity of HSCs and attenuates myeloid-biased differentiation in aged HSCs [66]. This effect is probably mediated by the mitochondrial unfolded protein response, a signaling pathway that regulates mitochondrial chaperone transcription and is critical for stress relief.
Age-related clonal hematopoiesis/clonal hematopoiesis of indeterminate potential
Recently, several groups have used next-generation genome sequencing to identify recurrent somatic mutations with clonal hematopoiesis in healthy elderly individuals [67,68,69]. This phenomenon is called age-related clonal hematopoiesis (ARCH) or clonal hematopoiesis of indeterminate potential (CHIP) [70, 71]. ARCH/CHIP determined using peripheral blood is extremely rare in young individuals (< 1% in persons < 40 years of age) but increases exponentially with age. These mutations are observed in 9.5% of the individuals aged 70 to 79, 11.7% of those aged 80 to 89, and 18.4% of those over 90 [68]. ARCH/CHIP is mainly associated with mutations in three epigenetic regulatory genes—DNMT3A, TET2, and ASXL1—that have been implicated in hematologic cancers and is a strong risk factor for subsequent hematological cancers [69] (Fig. 2a). Mutant HSCs in this spectrum acquire additional mutations that can lead to a disease phenotype and ultimately morbidity and mortality. Upon serial acquisition of mutations, ARCH/CHIP can progress to myelodysplastic syndrome (MDS) and ultimately acute myeloid leukemia (AML), directly to AML without an intervening MDS stage, or to other conditions such as myeloproliferative neoplasms (MPN) or lymphoid neoplasms [70, 72]. In addition to ARCH/CHIP, DNMT3A, TET2, and ASXL1 mutations are frequently detected in several myeloid malignancies, including MDS, MDS/MPN, and AML [73,74,75], suggesting that these mutations are the earliest events during malignant transformation.

Epigenetic regulators of clonal hematopoiesis. a Schematic representation of major epigenetic regulators involved in clonal hematopoiesis and age-associated myeloid malignancies. Mutation phenotypes of DNMT3A, TET2, and ASXL1 are summarized. 5-mC, 5-methylcytosine; 5-hmC, 5-hydroxymethylcytosine; H3K27me3, trimethylated H3 at lysine 27. b Age-associated clonal hematopoiesis. Age-related clonal hematopoiesis (ARCH)/clonal hematopoiesis of indeterminate potential (CHIP) is asymptomatic clonal hematopoiesis characterized mainly by mutations in TET2, DNMT3A, or ASXL1. ARCH/CHIP may progress with additional mutations such as RUNX1, EZH2, and IDH1/2 to MDS and ultimately AML. ARCH/CHIP may also progress directly to AML without an intervening MDS stage or to other conditions such as myeloproliferative neoplasms or lymphoid neoplasms. AML, acute myeloid leukemia; MDS, myelodysplastic syndrome; PRC, polycomb repressive complex
DNMT3A is a member of a family of DNA methyltransferases that catalyzes DNA methylation [76]. Loss-of-function mutations in Dnmt3a augment the self-renewal capacity of HSCs and cause myeloid-biased differentiation, leading to a fitness advantage in HSC clones in mice [77, 78]. Analysis of large adult AML cohorts revealed DNMT3A mutations frequently co-occurring with FLT3, NRAS, KRAS, PTPN11, and NPM1 [73, 74]. Experimental studies in mice confirmed that Dnmt3a loss synergized with an active Nras mutant, leading to the rapid development of leukemia [79]. Similar studies were performed with mutant Flt3 overexpression, which was shown to lead to the development of both myeloid and lymphoid leukemias [80].
TET2 is involved in DNA demethylation pathways converting 5-methylcytosine to 5-hydroxymethylcytosine (5-hmC), 5-formylcytosine, and 5-carboxylcytosine [81]. TET2 loss-of-function mutations are associated with hypermethylation. Mouse studies of conditional Tet2 loss revealed expansion of Lineage-Sca1+cKit+ cells concomitant with decreased 5-hmC levels [82, 83]. Tet2-mutant mice have been shown to synergize with Flt3-mutant mice, leading to the development of AML with full penetrance [84]. TET2 mutations frequently co-occur with loss-of-function EZH2 mutations in humans [85, 86]. We examined the impacts of loss-of-function EZH2 mutations on the pathogenesis of myeloid malignancies using Ezh2 conditional knockout mice [87] and demonstrated that an Ezh2 deficiency in combination with a Tet2 hypomorph in mice accelerates the transformation of HSCs and induces MDS and MDS/MPN [88].
ASXL1 is involved in mediating a number of histone modifications, such as H3K27me3, H2AK119Ub, and histone H3 at lysine 4 trimethylation (H3K4me3), which regulate gene expression, and might function as a scaffold for epigenetic regulators [89]. Loss of ASXL1 results in the global exclusion of H3K27me3, indicating that ASXL1 cooperates with PRC2 to regulate H3K27me3 [90]. ASXL1 forms a complex with the deubiquitination enzyme BAP1 and removes monoubiquitin from H2AK119Ub, to derepress genes targeted by PRC1 [91]. Recent studies using mice expressing an ASXL1 mutant demonstrated that an ASXL1 mutation alone is not sufficient for inducing the development of hematologic malignancies [92,93,94]. However, an ASXL1 mutation increased the susceptibility to leukemogenesis in concert with a RUNX1 mutant or in viral insertional mutagenesis, indicating that mice expressing an ASXL1 mutant represent a premalignant condition like ARCH/CHIP [93].
ARCH/CHIP progresses under selection pressure such as that imposed by aging, chemotherapy, or immune-mediated clonal selection [95]. Mutations in DNMT3A, TET2, and ASXL1 appear to offer a selective advantage to HSC clones over non-mutated clones by maximizing self-renewal and modulating differentiation, suggesting that a dysregulated epigenome increases the epigenetic heterogeneity that eventually leads to the appearance of HSC clones with better fitness in the aged BM niche. Recent studies have shown that chronic infection depletes normal HSCs and multipotent progenitor cells in an interferon γ-dependent manner [96] and that elevated levels of TNF selectively favor the expansion of TET2-mutant HSCs [97]. Moreover, Tet2 recruits Hdac2 and represses the transcription of Il6 via histone deacetylation [98]. Under inflammatory stress, Tet2-deficient HSCs resist apoptosis, rapidly expand, and produce more proinflammatory cytokines, including IL-6 [99]. These findings suggest that inflammation and inflammatory aged BM microenvironment enhance the clonal expansion of HSCs with driver mutations over normal aged HSCs [100].
As we described above, the co-occurrence of mutations is important for the progression from ARCH/CHIP to MDS and AML [79, 80, 84, 88, 93]. Additionally, the order of mutation acquisition may be crucial for disease pathogenesis (Fig. 2b) [72]. Targeted mutational analysis of patients with de novo and secondary AML revealed that mutations in SRSF2, SF3B1, U2AF1, ZRSR2, ASXL1, EZH2, BCOR, or STAG2 were highly specific for secondary AML and occurred early in leukemogenesis [101]. Makishima et al. identified two classes of mutated genes by sequencing MDS and secondary AML samples: type 1 enriched in secondary AML compared with high-risk MDS (FLT3, PTPN11, WT1, IDH1, NPM1, IDH2, and NRAS) and type 2 enriched in high-risk compared with low-risk MDS (TP53, GATA2, KRAS, RUNX1, STAG2, ASXL1, ZRSR2, and TET2) [102]. Another sequencing study of serial samples from patients with secondary AML revealed that mutations in genes related to DNA methylation and splicing machinery were early disease events of myelodysplasia [103]. In contrast, somatic variants associated with signaling pathways arose or their allelic burdens expanded during progression. Further studies of a larger number of serial samples may improve the understanding of the order of mutation acquisition.
Continuous follow-up is important for patients diagnosed with ARCH/CHIP. ARCH/CHIP can be transferred from donor to recipient during allogeneic HSC transplantation. Gondek et al. reported two cases of donor cell leukemia arising from ARCH/CHIP marked by somatic mutations in leukemia-related genes in donors [104]. This study suggests that the recognition and diagnosis of ARCH/CHIP are important before the assessment and selection of a donor, particularly for donors older than 50–60 years [105]. In contrast, screening for ARCH/CHIP among potential donors may have several ethical and operational implications related to disclosure of the results [106]. The current level of evidence is insufficient to support the implementation of prospective screening of donors for ARCH/CHIP. Further studies may reveal the magnitude of risk or benefit associated with screening for ARCH/CHIP.
ARCH/CHIP also has an impact on the development of non-malignant diseases. Coronary heart disease has been observed to occur 1.9 times more frequently in individuals with ARCH/CHIP than in those without ARCH/CHIP [107]. Although the mechanism underlying the association of ARCH/CHIP with coronary heart disease remains to be investigated, driver mutations such as TET2 and DNMT3A have been reported to cause phenotypic changes in HSCs and immune cells, including increased inflammatory responses in macrophages and mast cells, and functional alterations in T cells [107,108,109].
Application of single-cell technologies to HSC aging
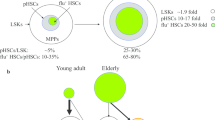
Recently, single-cell analysis has become a powerful tool for studying cellular differentiation pathways [110]. These advances in technology have made it possible to reveal differences intrinsic to the cell during the aging of HSCs. Grover et al. analyzed old and young HSC transcriptomes at the single-cell level and found that platelet-biased HSCs preferentially expand during aging, and aged HSCs are functionally platelet-biased [5]. They observed that loss of the FOG-1 transcription factor that is associated with HSC platelet priming increases lymphoid output, suggesting that increased platelet bias contributes to the age-associated decrease in lymphopoiesis. Yamamoto et al. performed large-scale clonal analysis using single HSC transplantation and found that the frequency of myeloid-restricted repopulating progenitors increases dramatically with age, while multipotent HSCs expand modestly in the BM [111]. They also identified a subset of functional HSCs that are myeloid-restricted in primary recipients but display multi-lineage outputs in secondary recipients.
Single-cell epigenomic technologies such as single-cell chromatin immunoprecipitation sequencing and single-cell assays for transposase-accessible chromatin (ATAC) sequencing have been employed to investigate histone modifications and to map the accessible chromatin regions, respectively [112]. Florian et al. performed a comprehensive set of paired daughter cell analyses that included single-cell 3D confocal imaging, single-cell transplants, single-cell RNA-seq, and single-cell ATAC-seq. They found that polar HSCs (that are the major cells in young mice) preferentially sort the small RhoGTPase Cdc42 asymmetrically while apolar HSCs (that are the majority in aged mice) sort symmetrically during cell division [113]. These findings suggest that the outcome of HSC division is strongly linked to the polarity status before mitosis, which is in turn determined by Cdc42 activity in HSCs. They also showed that the hematopoietic potential of daughter cells is linked to the amount of the epigenetic mark H4K16ac and to the amount of open chromatin allocated to a daughter cell, but not to its transcriptome. Cheung et al. employed a highly multiplexed mass cytometric analysis to profile the global levels of a wide array of chromatin modifications in immune cells at the single-cell level and found markedly different cell type- and hematopoietic lineage-specific chromatin modification patterns [114]. Differential analysis of HSPCs from younger and older adults revealed that aging was associated with increased heterogeneity between individuals and elevated cell-to-cell variability in chromatin modifications.
Conclusions
In this review, we have described various stresses that promote HSC aging and clonal hematopoiesis with age. We also discussed recent studies on HSC aging using single-cell technologies at both the transcriptomic and epigenetic levels. There is no doubt that HSCs show declining function during aging, but we still do not know whether this dysfunction is reversible in humans. Some promising strategies have been employed to rejuvenate aged HSCs based on stress factors intrinsic and extrinsic to the cell, including NAC and p38 MAPK inhibitor [16], CASIN [28], epigenetic manipulation such as Sirt3 and Sirt7 overexpression [65, 66], sympathomimetic supplementation [44], and ABT263 (navitoclax) [48] (Fig. 1). In humans, ABT263 has already been used, but its use has been limited by on-target thrombocytopenia [115, 116]. Nevertheless, such interventions may be adapted for treating HSC aging in humans with acceptable toxicity through new approaches in the formulation, delivery, or administration schedules.
Although the aging of HSCs has been extensively studied, there are some conflicting results. Therefore, we should carefully consider the differences in systems and experimental methods between these studies. We also need to examine how findings obtained from mice with 2-year lifespans can be extrapolated to human HSCs. The elderly population is rapidly increasing worldwide. Basic and clinical studies are needed to elucidate aging-related hematopoietic pathology to improve the clinical outcome of patients with these pathologies.
Availability of data and materials
Not applicable
Abbreviations
- AML:
-
Acute myeloid leukemia
- ARCH:
-
Age-related clonal hematopoiesis
- ATAC:
-
Assays for transposase-accessible chromatin
- BM:
-
Bone marrow
- CHIP:
-
Clonal hematopoiesis of indeterminate potential
- H2AK119Ub:
-
H2A at lysine 119 ubiquitination
- H3K4me3:
-
H3 at lysine 4 trimethylation
- H3K27me3:
-
H3 at lysine 27 trimethylation
- H4K16ac:
-
H4 at lysine 16 acetylation
- HSCs:
-
Hematopoietic stem cells
- HSPCs:
-
Hematopoietic stem and progenitor cells
- MAPK:
-
Mitogen-activated protein kinase
- MDS:
-
Myelodysplastic syndrome
- MPN:
-
Myeloproliferative neoplasms
- NAC:
-
N-acetyl cysteine
- NER:
-
Nucleotide excision repair
- NHEJ:
-
Non-homologous end joining
- PcG:
-
Polycomb group
- PRC:
-
Polycomb repressive complex
- ROS:
-
Reactive oxygen species
- SASP:
-
Senescence-associated secretory phenotype
- SNS:
-
Sympathetic nervous system
References
Dykstra B, Olthof S, Schreuder J, Ritsema M, de Haan G. Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J Exp Med. 2011;208(13):2691–703.
Sudo K, Ema H, Morita Y, Nakauchi H. Age-associated characteristics of murine hematopoietic stem cells. J Exp Med. 2000;192(9):1273–80.
Beerman I, Bhattacharya D, Zandi S, Sigvardsson M, Weissman IL, Bryder D, Rossi DJ. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc Natl Acad Sci U S A. 2010;107(12):5465–70.
Liang Y, Van Zant G, Szilvassy SJ. Effects of aging on the homing and engraftment of murine hematopoietic stem and progenitor cells. Blood. 2005;106(4):1479–87.
Grover A, Sanjuan-Pla A, Thongjuea S, Carrelha J, Giustacchini A, Gambardella A, Macaulay I, Mancini E, Luis TC, Mead A, et al. Single-cell RNA sequencing reveals molecular and functional platelet bias of aged haematopoietic stem cells. Nat Commun. 2016;7:11075.
Rundberg Nilsson A, Soneji S, Adolfsson S, Bryder D, Pronk CJ. Human and murine hematopoietic stem cell aging is associated with functional impairments and intrinsic megakaryocytic/erythroid bias. PLoS One. 2016;11(7):e0158369.
Juliusson G, Antunovic P, Derolf A, Lehmann S, Mollgard L, Stockelberg D, Tidefelt U, Wahlin A, Hoglund M. Age and acute myeloid leukemia: real world data on decision to treat and outcomes from the Swedish Acute Leukemia Registry. Blood. 2009;113(18):4179–87.
Verovskaya EV, Dellorusso PV, Passegue E. Losing sense of self and surroundings: hematopoietic stem cell aging and leukemic transformation. Trends Mol Med. 2019;25(6):494–515.
Akunuru S, Geiger H. Aging, clonality, and rejuvenation of hematopoietic stem cells. Trends Mol Med. 2016;22(8):701–12.
de Haan G, Lazare SS. Aging of hematopoietic stem cells. Blood. 2018;131(5):479–87.
Ermolaeva M, Neri F, Ori A, Rudolph KL. Cellular and epigenetic drivers of stem cell ageing. Nat Rev Mol Cell Biol. 2018;19(9):594–610.
Li X, Zeng X, Xu Y, Wang B, Zhao Y, Lai X, Qian P, Huang H. Mechanisms and rejuvenation strategies for aged hematopoietic stem cells. J Hematol Oncol. 2020;13(1):31.
Mejia-Ramirez E, Florian MC. Understanding intrinsic hematopoietic stem cell aging. Haematologica. 2020;105(1):22–37.
Naka K, Muraguchi T, Hoshii T, Hirao A. Regulation of reactive oxygen species and genomic stability in hematopoietic stem cells. Antioxid Redox Signal. 2008;10(11):1883–94.
Jang YY, Sharkis SJ. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood. 2007;110(8):3056–63.
Tothova Z, Kollipara R, Huntly BJ, Lee BH, Castrillon DH, Cullen DE, McDowell EP, Lazo-Kallanian S, Williams IR, Sears C, et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128(2):325–39.
Miyamoto K, Araki KY, Naka K, Arai F, Takubo K, Yamazaki S, Matsuoka S, Miyamoto T, Ito K, Ohmura M, et al. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell. 2007;1(1):101–12.
Wogan GN, Hecht SS, Felton JS, Conney AH, Loeb LA. Environmental and chemical carcinogenesis. Semin Cancer Biol. 2004;14(6):473–86.
Ju Z, Jiang H, Jaworski M, Rathinam C, Gompf A, Klein C, Trumpp A, Rudolph KL. Telomere dysfunction induces environmental alterations limiting hematopoietic stem cell function and engraftment. Nat Med. 2007;13(6):742–7.
Alter BP, Rosenberg PS, Giri N, Baerlocher GM, Lansdorp PM, Savage SA. Telomere length is associated with disease severity and declines with age in dyskeratosis congenita. Haematologica. 2012;97(3):353–9.
Rossi DJ, Bryder D, Seita J, Nussenzweig A, Hoeijmakers J, Weissman IL. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature. 2007;447(7145):725–9.
Flach J, Bakker ST, Mohrin M, Conroy PC, Pietras EM, Reynaud D, Alvarez S, Diolaiti ME, Ugarte F, Forsberg EC, et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature. 2014;512(7513):198–202.
Chambers SM, Shaw CA, Gatza C, Fisk CJ, Donehower LA, Goodell MA. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol. 2007;5(8):e201.
Iliakis G, Wang H, Perrault AR, Boecker W, Rosidi B, Windhofer F, Wu W, Guan J, Terzoudi G, Pantelias G. Mechanisms of DNA double strand break repair and chromosome aberration formation. Cytogenet Genome Res. 2004;104(1-4):14–20.
Mohrin M, Bourke E, Alexander D, Warr MR, Barry-Holson K, Le Beau MM, Morrison CG, Passegue E. Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis. Cell Stem Cell. 2010;7(2):174–85.
Kohler A, Schmithorst V, Filippi MD, Ryan MA, Daria D, Gunzer M, Geiger H. Altered cellular dynamics and endosteal location of aged early hematopoietic progenitor cells revealed by time-lapse intravital imaging in long bones. Blood. 2009;114(2):290–8.
Florian MC, Dorr K, Niebel A, Daria D, Schrezenmeier H, Rojewski M, Filippi MD, Hasenberg A, Gunzer M, Scharffetter-Kochanek K, et al. Cdc42 activity regulates hematopoietic stem cell aging and rejuvenation. Cell Stem Cell. 2012;10(5):520–30.
Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149(6):1192–205.
Famili F, Naber BA, Vloemans S, de Haas EF, Tiemessen MM, Staal FJ. Discrete roles of canonical and non-canonical Wnt signaling in hematopoiesis and lymphopoiesis. Cell Death Dis. 2015;6:e1981.
Florian MC, Nattamai KJ, Dorr K, Marka G, Uberle B, Vas V, Eckl C, Andra I, Schiemann M, Oostendorp RA, et al. A canonical to non-canonical Wnt signalling switch in haematopoietic stem-cell ageing. Nature. 2013;503(7476):392–6.
Povinelli BJ, Nemeth MJ. Wnt5a regulates hematopoietic stem cell proliferation and repopulation through the Ryk receptor. Stem Cells. 2014;32(1):105–15.
Krensky AM, Ahn YT. Mechanisms of disease: regulation of RANTES (CCL5) in renal disease. Nat Clin Pract Nephrol. 2007;3(3):164–70.
Ergen AV, Boles NC, Goodell MA. Rantes/Ccl5 influences hematopoietic stem cell subtypes and causes myeloid skewing. Blood. 2012;119(11):2500–9.
Yamaguchi A, Owen MJ, Takahashi S, et al. Maf promotes osteoblast differentiation in mice by mediating the age-related switch in mesenchymal cell differentiation. J Clin Invest. 2010;120(10):3455–65.
Singh L, Brennan TA, Russell E, Kim JH, Chen Q, Brad Johnson F, Pignolo RJ. Aging alters bone-fat reciprocity by shifting in vivo mesenchymal precursor cell fate towards an adipogenic lineage. Bone. 2016;85:29–36.
Naveiras O, Nardi V, Wenzel PL, Hauschka PV, Fahey F, Daley GQ. Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature. 2009;460(7252):259–63.
Ambrosi TH, Scialdone A, Graja A, Gohlke S, Jank AM, Bocian C, Woelk L, Fan H, Logan DW, Schurmann A, et al. Adipocyte accumulation in the bone marrow during obesity and aging impairs stem cell-based hematopoietic and bone regeneration. Cell Stem Cell. 2017;20(6):771–84 e776.
Kato Y, Hou LB, Miyagi S, Nitta E, Aoyama K, Shinoda D, Yamazaki S, Kuribayashi W, Isshiki Y, Koide S, et al. Bmi1 restricts the adipogenic differentiation of bone marrow stromal cells to maintain the integrity of the hematopoietic stem cell niche. Exp Hematol. 2019;76:24–37.
Kusumbe AP, Ramasamy SK, Adams RH. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature. 2014;507(7492):323–8.
Kusumbe AP, Ramasamy SK, Itkin T, Mae MA, Langen UH, Betsholtz C, Lapidot T, Adams RH. Age-dependent modulation of vascular niches for haematopoietic stem cells. Nature. 2016;532(7599):380–4.
Poulos MG, Ramalingam P, Gutkin MC, Llanos P, Gilleran K, Rabbany SY, Butler JM. Endothelial transplantation rejuvenates aged hematopoietic stem cell function. J Clin Invest. 2017;127(11):4163–78.
Katayama Y, Battista M, Kao WM, Hidalgo A, Peired AJ, Thomas SA, Frenette PS. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell. 2006;124(2):407–21.
Mendez-Ferrer S, Lucas D, Battista M, Frenette PS. Haematopoietic stem cell release is regulated by circadian oscillations. Nature. 2008;452(7186):442–7.
Maryanovich M, Zahalka AH, Pierce H, Pinho S, Nakahara F, Asada N, Wei Q, Wang X, Ciero P, Xu J, et al. Adrenergic nerve degeneration in bone marrow drives aging of the hematopoietic stem cell niche. Nat Med. 2018;24(6):782–91.
Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med. 2015;21(12):1424–35.
Gil J, Peters G. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol. 2006;7(9):667–77.
Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, Cheng T, DePinho RA, Sharpless NE, Scadden DT. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006;443(7110):421–6.
Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, Janakiraman K, Sharpless NE, Ding S, Feng W, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med. 2016;22(1):78–83.
Oshima M, Iwama A. Epigenetics of hematopoietic stem cell aging and disease. Int J Hematol. 2014;100(4):326–34.
Schuettengruber B, Bourbon HM, Di Croce L, Cavalli G. Genome regulation by polycomb and trithorax: 70 years and counting. Cell. 2017;171(1):34–57.
Park IK, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL, Morrison SJ, Clarke MF. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423(6937):302–5.
Lessard J, Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 2003;423(6937):255–60.
Iwama A, Oguro H, Negishi M, Kato Y, Morita Y, Tsukui H, Ema H, Kamijo T, Katoh-Fukui Y, Koseki H, et al. Enhanced self-renewal of hematopoietic stem cells mediated by the polycomb gene product Bmi-1. Immunity. 2004;21(6):843–51.
Hidalgo I, Herrera-Merchan A, Ligos JM, Carramolino L, Nunez J, Martinez F, Dominguez O, Torres M, Gonzalez S. Ezh1 is required for hematopoietic stem cell maintenance and prevents senescence-like cell cycle arrest. Cell Stem Cell. 2012;11(5):649–62.
Xie H, Xu J, Hsu JH, Nguyen M, Fujiwara Y, Peng C, Orkin SH. Polycomb repressive complex 2 regulates normal hematopoietic stem cell function in a developmental-stage-specific manner. Cell Stem Cell. 2014;14(1):68–80.
Kamminga LM, Bystrykh LV, de Boer A, Houwer S, Douma J, Weersing E, Dontje B, de Haan G. The Polycomb group gene Ezh2 prevents hematopoietic stem cell exhaustion. Blood. 2006;107(5):2170–9.
Konuma T, Nakamura S, Miyagi S, Negishi M, Chiba T, Oguro H, Yuan J, Mochizuki-Kashio M, Ichikawa H, Miyoshi H, et al. Forced expression of the histone demethylase Fbxl10 maintains self-renewing hematopoietic stem cells. Exp Hematol. 2011;39(6):697–709 e695.
Nakamura S, Oshima M, Yuan J, Saraya A, Miyagi S, Konuma T, Yamazaki S, Osawa M, Nakauchi H, Koseki H, et al. Bmi1 confers resistance to oxidative stress on hematopoietic stem cells. PLoS One. 2012;7(5):e36209.
Klauke K, Radulovic V, Broekhuis M, Weersing E, Zwart E, Olthof S, Ritsema M, Bruggeman S, Wu X, Helin K, et al. Polycomb Cbx family members mediate the balance between haematopoietic stem cell self-renewal and differentiation. Nat Cell Biol. 2013;15(4):353–62.
Nitta E, Itokawa N, Yabata S, Koide S, Hou LB, Oshima M, Aoyama K, Saraya A, Iwama A. Bmi1 counteracts hematopoietic stem cell aging by repressing target genes and enforcing the stem cell gene signature. Biochem Biophys Res Commun. 2020;521(3):612–9.
Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014;24(8):464–71.
Guarente L. Calorie restriction and sirtuins revisited. Genes Dev. 2013;27(19):2072–85.
Singh SK, Williams CA, Klarmann K, Burkett SS, Keller JR, Oberdoerffer P. Sirt1 ablation promotes stress-induced loss of epigenetic and genomic hematopoietic stem and progenitor cell maintenance. J Exp Med. 2013;210(5):987–1001.
Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, Tanokura M, Denu JM, Prolla TA. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010;143(5):802–12.
Brown K, Xie S, Qiu X, Mohrin M, Shin J, Liu Y, Zhang D, Scadden DT, Chen D. SIRT3 reverses aging-associated degeneration. Cell Rep. 2013;3(2):319–27.
Mohrin M, Shin J, Liu Y, Brown K, Luo H, Xi Y, Haynes CM, Chen D. Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science. 2015;347(6228):1374–7.
Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, McMichael JF, Schmidt HK, Yellapantula V, Miller CA, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20(12):1472–8.
Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488–98.
Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371(26):2477–87.
Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, Ebert BL. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126(1):9–16.
Jaiswal S, Ebert BL. Clonal hematopoiesis in human aging and disease. Science. 2019;366:6465.
Menssen AJ, Walter MJ. Genetics of progression from MDS to secondary leukemia. Blood. 2020;136(1):50–60.
Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, Hoadley K, Triche TJ Jr, Laird PW, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–74.
Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, Potter NE, Heuser M, Thol F, Bolli N, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209–21.
Ogawa S. Genetics of MDS. Blood. 2019;133(10):1049–59.
Okano M, Xie S, Li E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet. 1998;19(3):219–20.
Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, Bock C, Vasanthakumar A, Gu H, Xi Y, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. 2011;44(1):23–31.
Challen GA, Sun D, Mayle A, Jeong M, Luo M, Rodriguez B, Mallaney C, Celik H, Yang L, Xia Z, et al. Dnmt3a and Dnmt3b have overlapping and distinct functions in hematopoietic stem cells. Cell Stem Cell. 2014;15(3):350–64.
Mayle A, Yang L, Rodriguez B, Zhou T, Chang E, Curry CV, Challen GA, Li W, Wheeler D, Rebel VI, et al. Dnmt3a loss predisposes murine hematopoietic stem cells to malignant transformation. Blood. 2015;125(4):629–38.
Yang L, Rodriguez B, Mayle A, Park HJ, Lin X, Luo M, Jeong M, Curry CV, Kim SB, Ruau D, et al. DNMT3A loss drives enhancer hypomethylation in FLT3-ITD-associated leukemias. Cancer Cell. 2016;29(6):922–34.
Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333(6047):1300–3.
Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, Figueroa ME, Vasanthakumar A, Patel J, Zhao X, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20(1):11–24.
Quivoron C, Couronne L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, Do Cruzeiro M, Delhommeau F, Arnulf B, Stern MH, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell. 2011;20(1):25–38.
Shih AH, Jiang Y, Meydan C, Shank K, Pandey S, Barreyro L, Antony-Debre I, Viale A, Socci N, Sun Y, et al. Mutational cooperativity linked to combinatorial epigenetic gain of function in acute myeloid leukemia. Cancer Cell. 2015;27(4):502–15.
Makishima H, Jankowska AM, Tiu RV, Szpurka H, Sugimoto Y, Hu Z, Saunthararajah Y, Guinta K, Keddache MA, Putnam P, et al. Novel homo- and hemizygous mutations in EZH2 in myeloid malignancies. Leukemia. 2010;24(10):1799–804.
Khan SN, Jankowska AM, Mahfouz R, Dunbar AJ, Sugimoto Y, Hosono N, Hu Z, Cheriyath V, Vatolin S, Przychodzen B, et al. Multiple mechanisms deregulate EZH2 and histone H3 lysine 27 epigenetic changes in myeloid malignancies. Leukemia. 2013;27(6):1301–9.
Iwama A. Polycomb repressive complexes in hematological malignancies. Blood. 2017;130(1):23–9.
Muto T, Sashida G, Oshima M, Wendt GR, Mochizuki-Kashio M, Nagata Y, Sanada M, Miyagi S, Saraya A, Kamio A, et al. Concurrent loss of Ezh2 and Tet2 cooperates in the pathogenesis of myelodysplastic disorders. J Exp Med. 2013;210(12):2627–39.
Fujino T, Kitamura T. ASXL1 mutation in clonal hematopoiesis. Exp Hematol. 2020;83:74–84.
Abdel-Wahab O, Adli M, LaFave LM, Gao J, Hricik T, Shih AH, Pandey S, Patel JP, Chung YR, Koche R, et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell. 2012;22(2):180–93.
Scheuermann JC, de Ayala Alonso AG, Oktaba K, Ly-Hartig N, McGinty RK, Fraterman S, Wilm M, Muir TW, Muller J. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature. 2010;465(7295):243–7.
Hsu YC, Chiu YC, Lin CC, Kuo YY, Hou HA, Tzeng YS, Kao CJ, Chuang PH, Tseng MH, Hsiao TH, et al. The distinct biological implications of Asxl1 mutation and its roles in leukemogenesis revealed by a knock-in mouse model. J Hematol Oncol. 2017;10(1):139.
Nagase R, Inoue D, Pastore A, Fujino T, Hou HA, Yamasaki N, Goyama S, Saika M, Kanai A, Sera Y, et al. Expression of mutant Asxl1 perturbs hematopoiesis and promotes susceptibility to leukemic transformation. J Exp Med. 2018;215(6):1729–47.
Uni M, Masamoto Y, Sato T, Kamikubo Y, Arai S, Hara E, Kurokawa M. Modeling ASXL1 mutation revealed impaired hematopoiesis caused by derepression of p16Ink4a through aberrant PRC1-mediated histone modification. Leukemia. 2019;33(1):191–204.
Bowman RL, Busque L, Levine RL. Clonal hematopoiesis and evolution to hematopoietic malignancies. Cell Stem Cell. 2018;22(2):157–70.
Matatall KA, Jeong M, Chen S, Sun D, Chen F, Mo Q, Kimmel M, King KY. Chronic infection depletes hematopoietic stem cells through stress-induced terminal differentiation. Cell Rep. 2016;17(10):2584–95.
Abegunde SO, Buckstein R, Wells RA, Rauh MJ. An inflammatory environment containing TNFα favors Tet2-mutant clonal hematopoiesis. Exp Hematol. 2018;59:60–5.
Zhang Q, Zhao K, Shen Q, Han Y, Gu Y, Li X, Zhao D, Liu Y, Wang C, Zhang X, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature. 2015;525(7569):389–93.
Cai Z, Kotzin JJ, Ramdas B, Chen S, Nelanuthala S, Palam LR, Pandey R, Mali RS, Liu Y, Kelley MR, et al. Inhibition of inflammatory signaling in Tet2 mutant preleukemic cells mitigates stress-induced abnormalities and clonal hematopoiesis. Cell Stem Cell. 2018;23(6):833–49 e835.
Cook EK, Luo M, Rauh MJ. Clonal hematopoiesis and inflammation: partners in leukemogenesis and comorbidity. Exp Hematol. 2020;83:85–94.
Lindsley RC, Mar BG, Mazzola E, Grauman PV, Shareef S, Allen SL, Pigneux A, Wetzler M, Stuart RK, Erba HP, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015;125(9):1367–76.
Makishima H, Yoshizato T, Yoshida K, Sekeres MA, Radivoyevitch T, Suzuki H, Przychodzen B, Nagata Y, Meggendorfer M, Sanada M, et al. Dynamics of clonal evolution in myelodysplastic syndromes. Nat Genet. 2017;49(2):204–12.
Kim T, Tyndel MS, Kim HJ, Ahn JS, Choi SH, Park HJ, Kim YK, Yang DH, Lee JJ, Jung SH, et al. The clonal origins of leukemic progression of myelodysplasia. Leukemia. 2017;31(9):1928–35.
Gondek LP, Zheng G, Ghiaur G, DeZern AE, Matsui W, Yegnasubramanian S, Lin MT, Levis M, Eshleman JR, Varadhan R, et al. Donor cell leukemia arising from clonal hematopoiesis after bone marrow transplantation. Leukemia. 2016;30(9):1916–20.
DeZern AE, Gondek LP. Stem cell donors should be screened for CHIP. Blood Adv. 2020;4(4):784–8.
Gibson CJ, Lindsley RC. Stem cell donors should not be screened for clonal hematopoiesis. Blood Adv. 2020;4(4):789–92.
Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, McConkey M, Gupta N, Gabriel S, Ardissino D, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017;377(2):111–21.
Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, Wu CL, Sano S, Muralidharan S, Rius C, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017;355(6327):842–7.
Cull AH, Snetsinger B, Buckstein R, Wells RA, Rauh MJ. Tet2 restrains inflammatory gene expression in macrophages. Exp Hematol. 2017;55:56–70 e13.
Ye F, Huang W, Guo G. Studying hematopoiesis using single-cell technologies. J Hematol Oncol. 2017;10(1):27.
Yamamoto R, Wilkinson AC, Ooehara J, Lan X, Lai CY, Nakauchi Y, Pritchard JK, Nakauchi H. Large-scale clonal analysis resolves aging of the mouse hematopoietic stem cell compartment. Cell Stem Cell. 2018;22(4):600–7 e604.
Clark SJ, Lee HJ, Smallwood SA, Kelsey G, Reik W. Single-cell epigenomics: powerful new methods for understanding gene regulation and cell identity. Genome Biol. 2016;17:72.
Florian MC, Klose M, Sacma M, Jablanovic J, Knudson L, Nattamai KJ, Marka G, Vollmer A, Soller K, Sakk V, et al. Aging alters the epigenetic asymmetry of HSC division. PLoS Biol. 2018;16(9):e2003389.
Cheung P, Vallania F, Warsinske HC, Donato M, Schaffert S, Chang SE, Dvorak M, Dekker CL, Davis MM, Utz PJ, et al. Single-cell chromatin modification profiling reveals increased epigenetic variations with aging. Cell. 2018;173(6):1385–97 e1314.
Vogler M, Hamali HA, Sun XM, Bampton ET, Dinsdale D, Snowden RT, Dyer MJ, Goodall AH, Cohen GM. BCL2/BCL-X(L) inhibition induces apoptosis, disrupts cellular calcium homeostasis, and prevents platelet activation. Blood. 2011;117(26):7145–54.
He S, Sharpless NE. Senescence in health and disease. Cell. 2017;169(6):1000–11.
Acknowledgements
The authors are grateful to all staff at the Division of Stem Cell and Molecular Medicine, Center for Stem Cell Biology and Regenerative Medicine, The Institute of Medical Science, The University of Tokyo.
Funding
This work was supported by Grants-in-Aid for Scientific Research (#19H05653 and JP19J20503) and Scientific Research on Innovative Areas “Replication of Non-Genomic Codes” (#19H05746) from MEXT, Japan
Author information
Authors and Affiliations
Contributions
SK wrote the manuscript, and AI edited and finalized it. All authors approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
There is no competing interest to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kurosawa, S., Iwama, A. Aging and leukemic evolution of hematopoietic stem cells under various stress conditions. Inflamm Regener 40, 29 (2020). https://doi.org/10.1186/s41232-020-00138-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41232-020-00138-3