
Abstract
Leishmaniasis is an infectious disease transmitted by the sand fly. It is caused by over 20 different species of Leishmania and has affected over 14 million people worldwide. One of the main forms of control of leishmaniasis is chemotherapy, but this is limited by the high cost and/or toxicity of available drugs. We previously found three novel compounds with an iridoid tetracyclic skeleton to have activity against trypanosome parasites. In this study, we determined the activity of the three anti-trypanosome compounds against Leishmania using field strain, 010, and the lab strain Leishmania hertigi. The minimum inhibitory concentration (MIC) of the compounds against 010 was determined by microscopy while the IC50 of compounds against L. hertigi was determined by fluorescence-activated cell sorting with Guava viacount analysis. We found two of the three compounds, molucidin and ML-F52, to have anti-Leishmania activity against both strains. The fluor-microscope observation with DAPI stain revealed that both Molucidin and ML-F52 induced abnormal parasites with two sets of nucleus and kinetoplast in a cell, suggesting that compounds might inhibit cytokinesis in Leishmania parasites. Molucidin and ML-F52 might be good lead compounds for the development of new anti-Leishmania chemotherapy.
Similar content being viewed by others

Introduction
Leishmania is a protozoan parasite causing leishmaniasis. Parasites are transmitted by the sand fly, and an estimated 350 million people are at risk of infection in over 88 countries worldwide [1]. There are three forms of leishmaniasis: cutaneous, muco-cutaneous, and visceral leishmaniasis. The most common is cutaneous leishmaniasis, caused by over 20 different Leishmania spp. and characterized by ulcers or nodules on the skin. Muco-cutaneous leishmaniasis, which is caused by parasites of the Viannia subgenus, especially Leishmania (V.) braziliensis, Leishmania (V.) panamensis, and Leishmania (V.) guyanensis, and also Leishmania amazonensis, is characterized by a progressive destruction of the mucosa by ulcers that spread from the mouth and nose to the pharynx and larynx. The third form, visceral leishmaniasis which is caused by Leishmania donovani and Leishmania infantum, is a systemic infection characterized by prolonged fever, anemia, weight loss, hepatosplenomegaly, and, in severe cases, death [2–6]. In Ghana, one of the three ecological zones, arid Northern savanna, epidemiologically lies within the leishmaniasis belt in Africa. There have however been no reports of leishmaniasis in this region of the country. Most reported cases of leishmaniasis in Ghana have come from the Ho district of the Volta Region which is the moist semi-deciduous forest region of Ghana [7]. Cutaneous leishmaniasis (which may include muco-cutaneous, because of problems with diagnosis) is the only kind of leishmaniasis reported in this region. The species of Leishmania recorded in Ghana as the causative agent of cutaneous leishmaniasis is Leishmania major [8]. More recently, a new member of the Leishmania enriettii complex has been detected and isolated in Ghana [9].
The main form of control of leishmaniasis is chemotherapy. The use of the current anti-Leishmania drugs are however limited by high toxicity [10]. The treatment of leishmaniasis involves the use of specific anti-Leishmania drugs and the aggressive management of any bacteria or parasitic co-infection, hypovolemia (decreased blood volume), and malnutrition [10]. Pentavalent antimonials, the first line treatment for leishmaniasis, have an efficacy dependent on the species of Leishmania, the geographical location as well as the clinical presentation of the disease [11]. The antimonials have also been reported to have high toxicity that results in frequent and life-threatening side effects [12]. The second line of treatment, pentamidine and amphotericin B, is limited by high toxicity and/or emergence of resistance [13]. Like the antimonials, the second line of treatment requires parenteral administration. Last but not least, Miltefosine, the only oral treatment for leishmaniasis, is teratogenic and is therefore not suitable for women of childbearing age [14].
Although the promastigote stage of the Leishmania parasite is mainly found in the sand fly vector, their ease of handling has made them a very useful tool in the determination of the anti-Leishmania activity of compounds and extracts. Also, promastigotes have been reported to be a good option for the screening of compounds whose anti-Leishmania activity is not dependent of cell-mediated parasiticidal mechanisms [15]. This is because their activities against the promastigote stage would be representative of their activities against the intracellular amastigotes [15].
In recent times, there have been several reports on the use of medicinal plants for the treatment of various ailments. Medicinal plants like Zanthoxyum zanthoxyloides and Annona senegalensis have been reported to have some anti-Leishmania activity [16, 17]. In our previous study, we isolated three novel tetracyclic iridoid compounds from Morinda lucida benth, Molucidin, ML-2-3, and ML-F52 and found them to have anti-trypanosome activity [18]. Leishmania species belongs to the same kinetoplastid protozoan group with Trypanosoma and shares very similar life cycle and organelles such as flagellum and kinetoplast. In this study, we have tested those novel compounds against Leishmania spp. and found that two of these three compounds, Molucidin and ML-F52, had activities against Leishmania species, suggesting Molucidin and ML-F52 to be promising lead compounds for the treatment of leishmaniasis.
Materials and methods
Leishmania parasites
The field strain of L. enriettii (010) promastigotes, which had been isolated in Ghana, and the lab strain, Leishmania hertigi promastigotes, were used in this study because of their availability. Parasites were cultured in vitro according to conditions established previously [19]. Log phase of parasites (1 × 106 parasites/ml) was diluted to a parasitemia of 3 × 105 parasites/ml with M199 medium and used for the drug assay. Estimation of parasitemia was done with the Neubauer’s counting chamber.
In vitro viacount assay for L. hertigi and the Ghanaian field strain (010)
Leishmania promastigote forms of L. hertigi parasites were seeded and incubated in the presence or absence of (50–0.78 μM) compounds (dissolved in DMSO) for 48 h, after which the Guava reagent for viacount (Millipore) was added to the culture in the ratio of 1:1 and incubated for 30 min. Analysis was performed using the Millipore guava easyCyte 5HT (USA) according to the manufacture’s instruction.
Promastigotes of Leishmania 010 were seeded and incubated in the presence or absence of (50–0.78 μM) compounds for 96 h and the minimum inhibitory concentration (MIC) of the compounds determined by microscopy.
Fluor-microscopy analysis using DAPI
Parasites were incubated for 24 h under appropriate conditions (25 °C) with appropriate concentrations of active compounds, Molucidin and ML-F52. Parasites were then harvested and fixed with 4 % paraformaldehyde at room temperature for 5 min. Washing steps were carried out with PBS and PBST (0.1 % Triton X 100 in PBS) at room temperature for 5 min and 15 min, respectively. The parasites were stained with 4’,6-diamidino-2’-phenylindole dihydrochloride (DAPI) (5 μg/ml DAPI in PBS) for 10 min. After washing steps as above, the slides were mounted using parmafluor mounting reagent and covered with cover slips. The slides were observed under the Olympus fluorescent microscope (Olympus BX53) to detect any phenotypic changes in L. hertigi parasites.
Results and discussion
Activity of compounds against Leishmania parasites
The aim of the study was to investigate the anti-Leishmania properties of the three novel tetracyclic iridoid compounds previously found to have anti-trypanosome activity. In the determination of the effect of Molucidin, ML-2-3, and ML-F52 on L. hertigi and 010, parasites were challenged with 0–50 μM of each compound for 48 and 96 h to determine MIC and the concentration of compounds that inhibited parasite growth by 50 % (IC50) by microscopy and fluorescence-activated cell sorting (FACS) analysis, respectively. Molucidin and ML-F52 had anti-Leishmania activity against both strains while ML-2-3 had no activity. Molucidin showed anti-L. hertigi activity with IC50 of 4.24 μM and anti-010 activity with MIC of 4.167 μM (Table 1). Compound ML-F52 showed anti-L. hertigi activity with IC50 of 3.38 μM and anti-010 activity with MIC of 2.60 μM (Table 1). All three compounds share same side chains, iridoid tetracyclic skeleton, but differ in their functional group region. Molucidin and ML-F52 are esters while ML-2-3 is carboxylic acid. The functional groups may play a key role in the activity of the compounds against Leishmania. With respect to toxicity, ML-F52 had a selective index (SI values) in the range of 1.4–5.36 while Molucidin had SI values in the range of 1.67–2.20, respectively (Table 2).
Effect of compounds on parasite morphology and flagella function
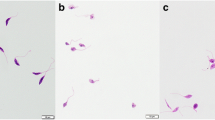
To investigate the effect of Molucidin and ML-F52 on the morphology and cell division of L. hertigi, we performed fluor-microscope observation with DAPI staining after incubation of parasite with 4 μM of Molucidin and ML-F52 for 48 h. Both Molucidin and ML-F52 induced short stumpy form parasite cells (Fig. 1a), in which two sets of nucleus and kinetoplast were observed (2N/2K), while normal cells had a set of nucleus and kinetoplast in each cell (1N/1K). Microscopic counting resulted in different ratios of both populations in Molucidin- and ML-F52-treated Leishmania as follows: normal cells (data not shown) and DMSO-treated cells showed 90 % of 1N/1K and 10 % of 2N/2K, while both Molucidin and ML-F52 caused significant increase with 30–40 % of 2N/2K and decrease of 1N/1K cells (Fig. 1b). These results indicated that Molucidin and ML-F52 might inhibit cytokinesis after division of both nuclei and kinetoplast in Leishmania parasites since there was also no observable inhibition of nuclear or kinetoplast division nor signs of nuclear and/or kinetoplast disintegration or fragmentation. This result indicated that Leishmania parasites challenged with both Molucidin and ML-F52 were able to undergo both nuclei and kinetoplast division but are unable to progress through mitosis to form two distinct daughter cells. The inability of cells to undergo cytokinesis has been shown to cause cell cycle arrest leading to the death of parasites [20, 21]. The inhibition of cytokinesis might imply the inability of cells to replicate in the mammalian host and increase the parasite burden. This may prevent the parasites from over running the host-immune and may give the host the edge to clear the infection [22]. On the other hand, this cell cycle inhibition may lead to the induction of apoptosis-like death in the Leishmania cells and may be the mechanism by which both compounds induce death [23, 24]. In fact, we previously found cell cycle alteration and apoptosis induction in Trypanosoma parasite challenged with ML-2-3 and ML-F52. Although there should be further analysis done to elucidate the mechanism of action for these compounds, they might be promising lead compounds for the development of an alternate anti-Leishmania drug.

Effect of Molucidin and ML-F52 on the nucleus, kinetoplast, and morphology of the parasite. a DAPI staining and fluor-microscope observation of parasites challenged with Molucidin and ML-F52. Both compounds induced short stumpy like parasite without long flagellum in which the cells having two sets of nucleus and kinetoplast in a cell were observed (indicated by red arrowhead). b Graph showed the ratio of normal cells which has one set of nucleus and kinetoplast (1N/1K) and abnormal cells induced by compounds which has two sets of nucleus and kinetoplast in a cell (2N/2K). 2N/2K cells were significantly induced by both Molucidin and ML-F52
Conclusion
Molucidin and ML-F52 are promising compounds for the development of novel anti-Leishmania drugs. The development of one or both compounds may lead to the development of a safer, cheaper, and relatively more available drug(s) for the treatment of leishmaniasis.
Abbreviations
FACS, fluorescence-activated cell sorting; DAPI, 4’,6-diamidino-2’-phenylindole dihydrochloride; MIC, minimum inhibitory concentration; IC50, compound concentration that inhibits parasite growth by 50 %; SI, selective index; N/K, nucleus and kinetoplast
References
Kimutai A, Ngure PK, Tonui WK, Gicheru MM. Review paper. J Infect Dis. 2009;3:14–25.
Herwaldt BL. Leishmaniasis. Lancet. 1999;354(9185):1191–9.
Lysenko AJ. Distribution of leishmaniasis in the Old World. Bull World Health Organ. 1971;44(4):515–20.
Murray HW, Berman JD, Davies CR, Saravia NG. Advances in leishmaniasis. Lancet. 2005;366:1561–77.
Alvar J, Yactayo S, Bern C. Leishmaniasis and poverty. Trends Parasitol. 2006;22(12):552–7.
Desjeux P. Leishmaniasis: current situation and new perspectives. Comp Immunol Microbiol Infect Dis. 2004;27(5):305–18.
Chappuis F, Sundar S, Hailu A, et al. Visceral leishmaniasis: what are the needs for diagnosis, treatment and control? Nat Rev Microbiol. 2007;5(11):873–82. doi:10.1038/nrmicro1748.
Boelaert M, Bhattacharya S. Evaluation of rapid diagnostic tests: visceral leishmaniasis. Nat Rev. 2007;5(11):S30-S39.
Kwakye-nuako G, Mosore M, Duplessis C, et al. First isolation of a new species of Leishmania responsible for human cutaneous leishmaniasis in Ghana and classification in the Leishmania enriettii complex. Int J Parasitol. 2015;45(11):679–84. doi:10.1016/j.ijpara.2015.05.001.
Guerin PJ, Olliaro P, Sundar S, et al. Visceral leishmaniasis: current status of control, diagnosis, and treatment, and a proposed research and development agenda. Lancet Infect Dis. 2002;2(8):494–501. doi:10.1016/S1473-3099(02)00347-X.
Pink R, Hudson A, Mouriès M-A, Bendig M. Opportunities and challenges in antiparasitic drug discovery. Nat Rev Drug Discov. 2005;4:727–40. doi:10.1038/nrd1824.
Singh OP, Sundar S. Immunotherapy and targeted therapies in treatment of visceral leishmaniasis: current status and future prospects. Front Immunol. 2014;5:1–9. doi:10.3389/fimmu.2014.00296.
Purkait B, Kumar A, Nandi N, et al. Mechanism of amphotericin B resistance in clinical isolates of Leishmania donovani. 2012:1031–41. doi:10.1128/AAC.00030-11.
Seifert K. Structures, targets and recent approaches in anti-leishmanial drug discovery and development. Open Med Chem J. 2011;5:31–9. doi:10.2174/1874104501105010031.
Vermeersch M, Inoce R, Tote K, Timmermans J, Cos P, Maes L. In vitro susceptibilities of Leishmania donovani promastigote and amastigote stages to antileishmanial reference drugs: practical relevance of stage-specific differences. Antimicrob Agents Chemother. 2009;53(9):3855–9. doi:10.1128/AAC.00548-09.
Maxim K, Loset J-R, Loset KN, et al. Anti-Leishmania activities associated with plants used in the Malian traditional medicine. J Ethnopharmacol. 2007;110(1):99-104
Mustapha AA. Annona senegalensis Persoon: a multipurpose shrub, its phytotherapic. Phytopharmacological and Phytomedicinal Uses. 2013;2(12):862–5.
Kwofie KD, Tung NH, Suzuki-Ohashi M, et al. In-vitro Anti-trypanosomal Activities and Mechanisms of Action of Novel Tetracyclic Iridoids from Morinda lucida Benth. Antimicrob Agents Chemother. 2016:AAC.01916-15. doi:10.1128/AAC.01916-15.
Mottram L. Protocols for handling and working with leishmania species (working with Leishmania for dummies); 2008.
Bompard G, Rabeharivelo G, Morin N. N-WASP / Arp2 / 3 complex pathway. 2008;14:1–14. doi:10.1186/1471-2121-9-42.
Bement WM. NIH Public Access. 2013;69(11):1010–20. doi:10.1002/cm.21085.Identification.
Castro EV, Yoneyama KG, Haapalainen EF, Toledo MS, Takahashi HK SA. Myriocin, a serine palmitoyltransferase inhibitor, blocks cytokinesis in Leishmania (Viannia) braziliensis promastigotes. Eukaryot Microbiol. 2013;60(4):377–87.
Das M, Mukherjee SB, Shaha C. Hydrogen peroxide induces apoptosis-like death in Leishmania donovani promastigotes. 2001.
Wheeler RJ, Gluenz E, Gull K. The cell cycle of Leishmania: morphogenetic events and their implications for parasite biology. 2011;79:647–62. doi:10.1111/j.1365-2958.2010.07479.x.
Acknowledgements
This study was supported by the “Japan Initiative for Global Research Network on Infectious Diseases (J-Grid)” program sponsored by the Japan Agency for Medical Research and Development (AMED).
Funding
The Japan Agency for Medical Research and Development (AMED) funded this study. The funding body did not play a role in the designing of the study, data collection, analysis, and interpretation and in the writing of the manuscript.
Availability of data and materials
The datasets during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors’ contributions
MAB performed, analyzed, and interpreted the parasitological data regarding screening and mechanistic studies and played the major roles in writing the manuscript. MTM, JA, KDK, GID, FAA, and SKB analyzed and interpreted the parasitological data regarding screening and mechanistic studies. WKA, IA, DAB, MO, and NO analyzed and interpreted all the data produced and played the major roles in writing the manuscript. KBAO, IT, PA, RAO, and TU performed, analyzed, and interpreted the toxicity data. NHT, FA, OM, AAA, and YS isolated and characterized the compounds and contributed to the writing of the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participant
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Amoa-Bosompem, M., Ohashi, M., Mosore, MT. et al. In vitro anti-Leishmania activity of tetracyclic iridoids from Morinda lucida, benth. Trop Med Health 44, 25 (2016). https://doi.org/10.1186/s41182-016-0026-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41182-016-0026-5