
Abstract
Background
It remains unclear whether hepatitis C virus (HCV)-infected Japanese patients on hemodialysis (HD) have an altered viral response to glecaprevir (GLE) and pibrentasvir (PIB) combination therapy and whether this treatment is safe for this patient group. Therefore, we investigated the characteristics associated with virological response to GLE and PIB combination therapy for HCV in Japanese patients undergoing HD. This study aimed to evaluate the safety and efficacy of GLE and PIB combination therapy in HCV-infected Japanese patients on HD.
Methods
This multicenter, single-arm, open-label, and prospective study was conducted at four centers in Japan and enrolled Japanese patients with chronic HCV genotype 1b, 2a, and 2b infections. Enrolled patients received 300 mg GLE in combination with 120 mg PIB once daily for either 8 or 12 weeks. The primary endpoint of this trial was the rate of sustained virological response at 12 weeks after treatment (SVR12). Furthermore, pharmacological effects and adverse events associated with combination therapy were evaluated.
Results
A total of 18 HD patients with HCV infection were enrolled in this study. Of these patients, 17 (94.4%) achieved SVR12. Among patients receiving combination therapy for 8 weeks, only one experienced a virological relapse at week 12 after the end of treatment. Despite the absence of treatment-related deaths, adverse events were observed in 9 patients (50.0%) during treatment. The most frequent adverse event was pruritus, which occurred in 7 patients (38.9%).
Conclusions
GLE and PIB combination therapy was effective and tolerable in Japanese HD patients with chronic HCV genotype 1b, 2a, and 2b infections.
Trial registration
This study was registered at the UMIN Clinical Trials Registry as UMIN000029781 (registered 21 November 2017, https://upload.umin.ac.jp/cgi-open-bin/ctr/ctr_view.cgi?recptno=R000034026).
Similar content being viewed by others


Background
The introduction of direct-acting antivirals (DAAs), which refer to small-molecule inhibitors of different viral proteins, has improved both the sustained virological response (SVR) rates and the tolerability of prolonged treatment in comparison with interferon (IFN)-based medicine [1, 2]. As treatment-related toxicity associated with IFN and ribavirin (RBV) is common, an IFN-free regimen with DAAs can be particularly effective for patients with end-stage renal disease, including those on hemodialysis (HD) [3,4,5].
In Japan, several combined regimens, including daclatasvir (DCV)/asunaprevir (ASV), ombitasvir/paritaprevir/ritonavir, and elbasvir/grazoprevir, which are primarily eliminated by hepatic metabolism, are available for the effective treatment of HD patients with hepatitis C virus (HCV) genotype 1b infections [6,7,8]. Despite the remarkable success of IFN-free regimens for patients with HCV genotype 1 infections, currently available regimens without RBV provide insufficient SVR rates in patients with genotype 2 infections.
Glecaprevir (GLE) is a nonstructural protein (NS) 3/4A protease inhibitor coformulated with pibrentasvir (PIB), an NS5A inhibitor, and is currently being investigated as a regimen with potential efficacy against all HCV genotypes [9,10,11]. The pharmacokinetics of GLE and PIB, which are primarily eliminated by hepatic metabolism, have been assessed in patients with end-stage renal disease. In addition, GLE and PIB are not removed from plasma by HD [12]. Recently, the EXPEDITION-4 trial in patients with HCV genotype 1–5 or 6 infection has reported the safety and efficacy of 12-week GLE and PIB combination therapy as treatment for chronic HCV in patients undergoing HD, showing dramatically improved rates of SVR at 12 weeks after treatment (SVR12) [12]. Therefore, this regimen is expected to yield beneficial results in specific populations, such as HCV genotype 2-infected Japanese patients with insufficient renal function. However, it remains unclear whether HCV-infected Japanese patients on HD experience positive outcomes with respect to SVR and safety following GLE and PIB combination therapy. Furthermore, the treatment duration of GLE and PIB combination therapy was 8 weeks for patients who neither had compensated liver cirrhosis nor had received previous DAA therapy in Japan. To the best of our knowledge, there are no data on the efficacy of 8-week GLE and PIB combination therapy in patients undergoing HD. Accordingly, this study aimed to evaluate the safety and efficacy of GLE and PIB combination therapy in HCV-infected Japanese patients on HD.
Methods
Ethics
This study was approved by the institutional review board (IRB)/ethics committee of Kitasato University School of Medicine (IRB number: C17-237) and Tokushukai Medical Group (IRB number: TGE00926-024) and is registered in the University Hospital Medical Information Network (UMIN) Clinical Trials Registry as UMIN000029781.
Study design and patients
This multicenter, single-arm, open-label, and prospective study was conducted at four centers in Japan. The enrollment commenced in November 2017, and the study was completed in July 2018. Patients with chronic HCV genotype 1b, 2a, and 2b infections who were undergoing HD received GLE and PIB. All enrolled patients received 300 mg GLE in combination with 120 mg PIB once daily for either 8 or 12 weeks. According to the manufacturer’s prescribing information for both medications, treatment duration should be 12 weeks for patients who had compensated liver cirrhosis and/or received previous DAA therapy. Diagnosis of liver cirrhosis was based on laboratory results, ultrasonography, and computed tomography revealing a hepatic cirrhotic appearance and/or splenomegaly. Liver biopsy was not performed for all patients because of the high risk of life-threatening complications. The discontinuation criteria for the enrolled patients were as follows: (1) occurrence of viral breakthrough (increase in plasma HCV-RNA levels exceeding 1 log10 IU/mL compared with the lowest recorded on-treatment value), (2) occurrence of severe adverse events according the National Cancer Institute’s Common Terminology Criteria for Adverse Events (CTCAE) version 4.0, and (3) patient’s desire to terminate.
Eligibility criteria
This study enrolled Japanese patients with chronic HCV genotype 1b, 2a, and 2b infections for at least 6 months and plasma HCV-RNA levels ≥ 2 log10 IU/mL. Treatment-naïve or treatment-experienced patients (previously treated with IFN-based therapy and/or IFN-free therapy) aged ≥ 20 years who were currently undergoing HD were included in the study. Conversely, patients with (1) decompensated liver cirrhosis (Child–Pugh classes B and C), (2) hepatocellular carcinoma, (3) resistance-associated variants of NS5A-P32 deletion among those with HCV genotype 1b, (4) infection/coinfection with hepatitis B virus or human immunodeficiency virus, (5) previous exposure to IFN-based therapy or DAA inhibitors within 1 month before drug administration, and (6) alanine aminotransferase (ALT) levels greater than five times the upper limit of the normal range, platelet and white blood cell (WBC) counts lower than 50,000 and 4000/mm3, respectively, and hemoglobin (Hb) levels less than 8.5 g/dL were excluded from the study.
Clinical parameters
The clinical characteristics of enrolled patients were evaluated. Data on demographics, plasma HCV-RNA levels, and baseline laboratory results before study drug administration were collected. Blood samples were obtained at each study visit before dialysis, and plasma HCV-RNA levels were quantified using Cobas TaqMan version 2.0 assay (Roche Diagnostics, Tokyo, Japan). The lower limit of quantification was 1.2 log10 IU/mL. HCV-RNA levels were measured at baseline; at treatment weeks 4, 8, and 12 (in patients in the 12-week treatment duration group); and at post-treatment weeks 4, 8, and 12.
We analyzed baseline resistance-associated variants at signature amino acid positions associated with resistance for genotype 1b using direct sequencing, such as NS5A-Y93, L31, P32, and NS3/4 D168 [13]. With respect to the laboratory tests performed, WBC, neutrophil, and platelet counts and levels of Hb, total protein, serum albumin, blood urea nitrogen (BUN), serum creatinine, total bilirubin, aspartate aminotransferase (AST), ALT, Mac-2-binding protein glycan isomer (M2BPGi), and α-fetoprotein were analyzed.
Efficacy
The primary endpoint of this trial was the SVR rate in the cohort (HCV-RNA level below the lower level of quantification) at 12 weeks after the end of treatment (SVR12) according to intention-to-treat analysis. We also investigated the mean decrease in plasma HCV-RNA levels between baseline and week 4 after combination therapy administration and the proportion of patients showing SVR at week 4.
Safety assessment
To evaluate safety and tolerability, we assessed the pharmacological effects and adverse events that occurred after the initiation of combination therapy during each study visit from the first day of study drug administration to 30 days after the last dose. The severity of any adverse events was graded using CTCAE version 4.0.
Statistical evaluation
Data were analyzed using the SPSS v.24.0 software package (IBM Corp., Armonk, NY, USA). All data were expressed as minimum, maximum, and median values. To assess the pharmacological effects of study treatment, Wilcoxon signed-rank test was used for the analysis of paired data. All differences with a p value < 0.05 were considered significant.
Results
Characteristics of patients
Among 1022 HD patients who were serologically tested in 16 dialysis facilities using third-generation enzyme-linked immunosorbent assay, 56 (5.5%) and 20 (2.0%) patients tested positive for hepatitis C antibody and HCV RNA, respectively. Of these 20 patients, two dropped out during the run-in period owing to failure to meet the criteria for commencing treatment, whereas the remaining 18 patients received combination therapy. Among these patients, two did not complete the study owing to clinical events (Fig. 1). Thirteen patients received combination therapy for 8 weeks, and five patients who had compensated liver cirrhosis and/or underwent previous DAA therapy received combination therapy for 12 weeks. The demographics and other baseline clinical characteristics of enrolled patients are summarized in Table 1.

Study flow diagram
The median age of patients was 67.5 years (range, 48–84 years), and 14 patients (77.8%) were male. Furthermore, four patients (22.2%) had liver cirrhosis, and four (22.2%) were previously treated with IFN-based and/or DAA-based therapy. Nine, six, and three patients were infected with genotypes 1b, 2a, and 2b, respectively. The HCV level was 6.55 log10 IU/mL (range, 4.1–7.3 log10 IU/mL). NS5A-Y93H, L31 M/V, and D168E in genotype 1b were detected in three patients (11.7%) who had a history of DAA therapy (DCV and ASV combination therapy). The duration of HD was 2.7 years (range, 0.3–30 years), and the cause of end-stage renal disease was diabetic nephropathy, hypertensive kidney disease, and glomerulonephritis in nine, five, and two patients, respectively. The serum albumin, serum creatinine, and ALT levels were 4.0 g/dL (range, 2.4–4.5 g/dL), 8.6 mg/dL (range, 4.4–13.0 mg/dL), and 16 IU/L (range, 5–168 IU/L), respectively.
Virological response
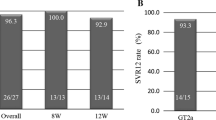
The results of the primary endpoint analysis indicated an SVR12 rate of 94.4% (17/18). The SVR12 rates in patients with genotype 1b, 2a, and 2b infections were 9/9, 6/6, and 2/3, respectively. The median decrease in plasma HCV-RNA levels between baseline and week 4 in all patients after combination therapy administration was 5.5 log10 IU/mL.
Patients receiving combination therapy for 8 weeks vs. 12 weeks
Among patients treated with combination therapy for 8 weeks, only one patient experienced virological relapse at week 12 after the end of treatment. The proportions of patients with undetectable HCV-RNA levels at treatment weeks 4 and 8 and at post-treatment week 12 were 100% (13/13), 100% (13/13), and 92.3% (12/13), respectively.
In patients treated with combination therapy for 12 weeks, including those with liver cirrhosis and/or previous DAA exposure, two clinical events led to the discontinuation of combination therapy. The proportions of patients with undetectable HCV-RNA levels at treatment weeks 4 and 8 and at post-treatment week 12 were 100% (5/5), 100% (4/4), and 100% (5/5), respectively.
Treatment response for previous DAA treatment failure
The demographics of patients with previous DAA treatment failure and other baseline clinical characteristics are summarized in Table 2. The previous DAA treatments for all patients were DCV and ASV combination therapy. All patients who experienced previous virological failure following DAA treatment substantially had variants within the HCV NS 3/4 and NS5A region. The SVR12 rate for patients who had previous DAA treatment failure was 100% (3/3).
Virological failure
Virological failure was observed in only one patient. After combination therapy administration for 8 weeks, virological relapse occurred at week 12 after the end of treatment. The patient was a 63-year-old man who had HCV genotype 2b and HCV-RNA level of 6.5 log10 IU/mL without a history of previous DAA exposure. The duration of HD was 6.5 years, and the cause of end-stage renal disease was diabetic nephropathy. Ultrasonography revealed chronic liver disease. The M2BPGi level was 0.6 C.O.I.; the AST to platelet ratio index and FIB-4 index were 1.667 and 1.03, respectively.
Pharmacological effects
Following drug administration, a decrease in the median ALT level from 16 to 8 IU/L and in the mean M2BPGi level from 1.8 to 1.5 C.O.I. was observed at the end of treatment (p < 0.001, 0.075, respectively). The baseline and post-treatment Hb levels were indistinguishable (11.8 vs. 11.6 g/dL, p = 0.757). There were no significant differences in WBC, neutrophil, and platelet counts and levels of total protein, serum albumin, BUN, serum creatinine, total bilirubin, and α-fetoprotein between baseline and post-treatment.
Safety assessment
The incidences of drug-related adverse events and adverse drug reactions are shown in Table 3. Adverse events were observed in nine patients (50.0%) during treatment. The most frequent adverse event was pruritus, which was noted in seven patients (38.9%). Specifically, pruritus occurred in 4 of 14 (28.6%) cases with chronic hepatitis and 3 of 4 (75%) cases with compensated liver cirrhosis. Pruritus occurred more frequently in patients with compensated liver cirrhosis. There were no grade 2 or higher abnormalities in AST, ALT, and total bilirubin levels, and grade 3 or higher anemia was not observed. Although there were no treatment-related deaths, two clinical events led to the discontinuation of combination therapy. One patient with a history of HCV treatment with DAA was hospitalized because of right leg cellulitis (grade 3) at treatment week 6. Cellulitis was not considered by the trial investigators to be drug-related. Another patient who had compensated liver cirrhosis exhibited grade 2 pruritus at week 2, and treatment with GLE and PIB was scheduled for 12 weeks. Although antihistamine and nalfurafine hydrochloride were administered, the patient opted to discontinue the treatment with these drugs at week 8.
Discussion
In a meta-analysis, Fabrizi et al. reported that the estimated adjusted relative risk of all-cause mortality was 1.34 times (95% confidence interval, 1.13–1.59) higher in anti-HCV-positive patients on HD than in anti-HCV-negative patients [14]. Söderholm et al. showed that IFN-based therapy improved the survival (odds ratio, 3.90) of patients with chronic hepatitis C infection before and during HD compared with that of untreated patients [15]. Therefore, the high therapeutic effect of DAAs may further improve the outcomes of HD patients.
A phase III trial has reported the efficacy and safety of GLE and PIB for HCV in 332 Japanese patients. Of these patients, 320 did not have renal impairment, and the SVR12 rate for patients without renal impairment was 97.8% (313/320) [16]. In this study, GLE and PIB combination therapy yielded an SVR rate of 94.4% for Japanese HD patients infected with HCV genotypes 1, 2a, and 2b. The present study showed that the SVR rate for patients with or without renal impairment was equivalent.
Until the introduction of GLE and PIB combination therapy, no IFN-free treatment without RBV for HCV genotype 2 infections was approved in Japan. DAA regimens with RBV for patients with stage 4 or 5 chronic kidney disease, including patients on HD, showed low tolerability and high rates of treatment discontinuation due to adverse events, such as reduced Hb level and elevated indirect bilirubin level [17,18,19]. GLE and PIB combination therapy, which can be provided regardless of baseline genotype, is often chosen because the drugs are cleared by the biliary system, and dose adjustment is not required for patients with severe renal impairment, including those on HD [20, 21]. This treatment regimen could contribute to the clinical practice in Japan, in which treatment options for patients with HCV genotype 2 infections who are undergoing HD remain few.
Our findings indicated that GLE and PIB combination therapy was effective in HD patients, including those with a history of DAA therapy and/or compensated liver cirrhosis. Phase I, II, and III trials on GLE and PIB combination therapy over an 8-week duration showed that a history of DAA therapy, genotype, and liver cirrhosis were independent factors affecting treatment response [22, 23]. In the present study, a treatment duration of 12 weeks led to positive outcomes in HD patients with liver cirrhosis and previous DAA treatment failure, suggesting that a sufficient treatment duration is viable in this patient group.
However, only one patient who received combination therapy for 8 weeks was reported to have had virological failure in this study. He had HCV genotype 2b without liver cirrhosis and a history of previous DAA therapy. The EXPEDITION-4 trial reported that no patients experienced virological failure with 12-week GLE and PIB combination therapy [12]. Although this patient had no risk factor for virological failure, to our knowledge, laboratory results and imaging examinations may have led to the underestimation of fibrosis stage. A treatment duration of 8 weeks may have been insufficient for this patient because of the extent and progression of liver fibrosis.
In this study, there were no treatment-related deaths or severe adverse events. A phase III trial also reported that GLE and PIB combination therapy was well tolerated by patients with no serious drug-related adverse events. A phase III trial in Japan reported a frequency rate of 6.9% (22/320) for pruritus in patients without renal impairment [16]. The frequency rate for pruritus (38.9%) in the present study was considerably higher than the rates in previous reports. Pruritus is a frequent side effect of these drugs, regardless of compensated liver cirrhosis. The mechanism for this remains unclear owing to limited available data. Moreover, pruritus is a symptom frequently observed in patients on HD, affecting 53.8% of patients on HD; however, its etiology is not completely understood [24]. Therefore, the incidence of pruritus may have increased because of the additive effect of GLE and PIB administration in patients undergoing dialysis.
In the present study, two patients who showed SVR12 had clinical events that led to the discontinuation of combination therapy. One patient with compensated liver cirrhosis chose to terminate treatment because of pruritus. Pruritus occurs more frequently in patients with compensated liver cirrhosis and can be more severe. Another patient with chronic cardiac disease was hospitalized owing to right leg cellulitis at treatment week 6. As such, cellulitis was likely due to edema resulting from the patient’s poor cardiac function. We determined that this clinical event was not caused by combination therapy. It is well known that HCV patients on dialysis are at an increased risk of treatment discontinuation owing to adverse events unrelated to HCV treatment, such as infection and/or cardiac disease. Therefore, a shorter treatment duration is often more beneficial in this patient group.
The present study has three limitations. First, the study was performed on a small sample of patients; therefore, there might have been some selection bias. Second, it is unclear whether DAAs improve the long-term prognoses of these patients. Finally, as previously mentioned, more detailed in vivo pharmacokinetic studies on GLE and PIB combination therapy should be performed to determine whether HD affects serum concentrations and drug activity in patients.
Conclusions
The results of this single-arm, open-label study indicated that an RBV-free coformulation of GLE and PIB is effective as treatment for chronic HCV genotype 1b, 2a, and 2b infections in Japanese patients undergoing HD.
Abbreviations
- ALT:
-
Alanine aminotransferase
- AST:
-
Aspartate aminotransferase
- ASV:
-
Asunaprevir
- BUN:
-
Blood urea nitrogen
- CTCAE:
-
Common Terminology Criteria for Adverse Events
- DAAs:
-
Direct-acting antivirals
- DCV:
-
Daclatasvir
- GLE:
-
Glecaprevir
- Hb:
-
Hemoglobin
- HCV:
-
Hepatitis C virus
- HD:
-
Hemodialysis
- IFN:
-
Interferon
- M2BPGi:
-
Mac-2-binding protein glycan isomer
- NS:
-
Nonstructural protein
- PIB:
-
Pibrentasvir
- RBV:
-
Ribavirin
- SVR:
-
Sustained virological response
- SVR12:
-
Sustained virological response at 12 weeks after treatment
- UMIN:
-
University Hospital Medical Information Network
- WBC:
-
White blood cells
References
Toyoda H, Atsukawa M, Takaguchi K, Senoh T, Michitaka K, Hiraoka A, et al. Real-world virological efficacy and safety of elbasvir and grazoprevir in patients with chronic hepatitis C virus genotype 1 infection in Japan. J Gastroenterol. 2018. https://doi.org/10.1007/s00535-018-1473-z.
Okubo T, Atsukawa M, Tsubota A, Toyoda H, Shimada N, Abe H, et al. Efficacy and safety of ledipasvir/sofosbuvir for genotype 1b chronic hepatitis C patients with moderate renal impairment. Hepatol Int. 2018;12:133–42.
Pol S, Jadoul M, Vallet-Pichard A. An update on the management of hepatitis C virus-infected patients with stage 4-5 chronic kidney disease while awaiting the revised KDIGO guidelines. Nephrol Dial Transplant. 2017;32:32–5.
Arai T, Atsukawa M, Tsubota A, Ikegami T, Shimada N, Kato K, et al. Efficacy and safety of ombitasvir/paritaprevir/ritonavir combination therapy for genotype 1b chronic hepatitis C patients complicated with chronic kidney disease. Hepatol Res. 2018;48:549–55.
Alseiari M, Meyer KB, Wong JB. Evidence underlying KDIGO (kidney disease: improving global outcomes) guideline recommendations: a systematic review. Am J Kidney Dis. 2016;67:417–22.
Uojima H, Kobayashi S, Hidaka H, Shuichi M, Takayasu O, Takeshi K, et al. Virological response to daclatasvir and asunaprevir combination therapy for chronic hepatitis C virus genotype 1b infection in dialysis patients: a prospective, multicenter study. Ren Replace Ther. 2017;3:7.
Morisawa N, Koshima Y, Kuriyama S, Matsuyama M, Hayashi N, Satoh JI, et al. Effectiveness of a fixed combination formula of ombitasvir/paritaprevir/ritonavir for hepatitis C virus infection in patients on maintenance haemodialysis. Nephrology. 2017;22:562–5.
Maunoury F, Clément A, Nwankwo C, Levy-Bachelot L, Abergel A, Di Martino V, et al. Cost-effectiveness analysis of elbasvir-grazoprevir regimen for treating hepatitis C virus genotype 1 infection in stage 4-5 chronic kidney disease patients in France. PLoS One. 2018;13:e0194329.
Kosloski MP, Zhao W, Marbury TC, Preston RA, Collins MG, Pugatch D, et al. Effects of renal impairment and hemodialysis on the pharmacokinetics and safety of the glecaprevir and pibrentasvir combination in hepatitis C virus-negative subjects. Antimicrob Agents Chemother. 2018;62:e01990–17.
Krishnan P, Schnell G, Tripathi R, Beyer J, Reisch T, Dekhtyar T, et al. Integrated resistance analysis of CERTAIN-1 and CERTAIN-2 studies in hepatitis C virus-infected patients receiving glecaprevir and pibrentasvir in Japan. Antimicrob Agents Chemother. 2018;62:e02217 17.
Poordad F, Pol S, Asatryan A, Buti M, Shaw D, Hézode C, et al. Glecaprevir/pibrentasvir in patients with hepatitis C virus genotype 1 or 4 and past direct-acting antiviral treatment failure. Hepatology. 2018;67:1253–60.
Gane E, Lawitz E, Pugatch D, Papatheodoridis G, Bräu N, Brown A, et al. Glecaprevir and pibrentasvir in patients with HCV and severe renal impairment. N Engl J Med. 2017;377:1448–55.
Uchida Y, Kouyama J, Naiki K, Mochida S. A novel simple assay system to quantify the percent HCV-RNA levels of NS5A Y93H mutant strains and Y93 wild-type strains relative to the total HCV-RNA levels to determine the indication for antiviral therapy with NS5A inhibitors. PLoS One. 2014;9:e112647.
Fabrizi F, Takkouche B, Lunghi G, Dixit V, Messa P, Martin P. The impact of hepatitis C virus infection on survival in dialysis patients: meta-analysis of observational studies. J Viral Hepat. 2007;14:697–703.
Söderholm J, Millbourn C, Büsch K, Kövamees J, Schvarcz R, Lindahl K, et al. Higher risk of renal disease in chronic hepatitis C patients: antiviral therapy survival benefit in patients on hemodialysis. J Hepatol. 2018;68:904–11.
Atsukawa M, Chayama K, Suzuki F, Sato K, Karino Y, Atarashi T, et al. Efficacy and safety of glecaprevir/pibrentasvir in patients infected with HCV GT1 - 3 by renal impairment status: a pooled analysis of two phase 3 Japanese trials. Hepatology. 2017;66:634A–5A.
Ozer Etik D, Ocal S, Boyacioglu AS. Hepatitis C infection in hemodialysis patients: a review. World J Hepatol. 2015;7:885–95.
Abad S, Vega A, Hernández E, Mérida E, de Sequera P, Albalate M, et al. Universal sustained viral response to the combination of ombitasvir/paritaprevir/ritonavir and dasabuvir with/without ribavirin in patients on hemodialysis infected with hepatitis C virus genotypes 1 and 4. Am J Nephrol. 2017;45:267–72.
Liu CH, Liu CJ, Huang CF, Lin JW, Dai CY, Liang CC, et al. Peginterferon alfa-2a with or without low-dose ribavirin for treatment-naive patients with hepatitis C virus genotype 2 receiving haemodialysis: a randomised trial. Gut. 2015;64:303–11.
Franczyk-Skóra B, Gluba-Brzózka A, Wranicz JK, Banach M, Olszewski R, Rysz J. Sudden cardiac death in CKD patients. Int Urol Nephrol. 2015;47:971–82.
Pockros PJ, Reddy KR, Mantry PS, Cohen E, Bennett M, Sulkowski MS, et al. Efficacy of direct-acting antiviral combination for patients with hepatitis C virus genotype 1 infection and severe renal impairment or end-stage renal disease. Gastroenterology. 2016;150:1590–8.
Puoti M, Foster GR, Wang S, Mutimer D, Gane E, Moreno C, et al. High SVR12 with 8-week and 12-week glecaprevir/pibrentasvir therapy: an integrated analysis of HCV genotype 1-6 patients without cirrhosis. J Hepatol. 2018;69:293–300. https://doi.org/10.1016/j.jhep.2018.03.007.
Gane E, Poordad F, Wang S, Asatryan A, Kwo PY, Lalezari J, et al. High efficacy of ABT-493 and ABT-530 treatment in patients with HCV genotype 1 or 3 infection and compensated cirrhosis. Gastroenterology. 2016;151:651–9.
Peres LAB, Passarini SR, Branco MFBT, Kruger LA. Skin lesions in chronic renal dialysis. J Bras Nefrol. 2014;36:42–7.
Acknowledgements
We thank Robert E. Brandt, Founder, CEO, and CME of MedEd Japan, for editing and formatting the manuscript. We also express our gratitude to Atsuko Takeuchi and Horio Kazue, laboratory technicians of Kitasato University School of Medicine, for analyzing resistance-associated variants.
Funding
This research received no funding.
Availability of data and materials
Data are uploaded as supporting information files.
Author information
Authors and Affiliations
Contributions
YT and HU contributed equally to this work. YT, TM, YT, and WK designed the research. TO, TF, YY, NW, KK, HH, TN, AS, TA, JS, and MM performed the research. HU analyzed the data. YT and HU wrote the manuscript. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the institutional review board (IRB)/ethics committee of Kitasato University School of Medicine (IRB number: C17-237) and Tokushukai Medical Group (IRB number: TGE00926-024). Informed consent was obtained from patients and their families before study participation commenced.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Tanaka, Y., Masaki, T., Uojima, H. et al. Glecaprevir and pibrentasvir combination therapy for hepatitis C virus-infected Japanese patients on hemodialysis. Ren Replace Ther 4, 51 (2018). https://doi.org/10.1186/s41100-018-0193-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41100-018-0193-4