
Abstract
Background
Uveal melanoma (UVM) stands as the predominant type of primary intraocular malignancy among adults. The clinical significance of N7-methylguanosine (m7G), a prevalent RNA modifications, in UVM remains unclear.
Methods
Primary information from 80 UVM patients were analyzed as the training set, incorporating clinical information, mutation annotations and mRNA expression obtained from The Cancer Genome Atlas (TCGA) website. The validation set was carried out using Gene Expression Omnibus (GEO) database GSE22138 and GSE84976. Kaplan–Meier and Cox regression of univariate analyses were subjected to identify m7G-related regulators as prognostic genes.
Result
A prognostic risk model comprising EIF4E2, NUDT16, SNUPN and WDR4 was established through Cox regression of LASSO. Evaluation of the model’s predictability for UVM patients’ prognosis by Receiver Operating Characteristic (ROC) curves in the training set, demonstrated excellent performance Area Under the Curve (AUC) > 0.75. The high-risk prognosis within the TCGA cohort exhibit a notable worse outcome. Additionally, an independent correlation between the risk score and overall survival (OS) among UVM patients were identified. External validation of this model was carried out using the validation sets (GSE22138 and GSE84976). Immune-related analysis revealed that patients with high score of m7G-related risk model exhibited elevated level of immune infiltration and immune checkpoint gene expression.
Conclusion
We have developed a risk prediction model based on four m7G-related regulators, facilitating effective estimate UVM patients’ survival by clinicians. Our findings shed novel light on essential role of m7G-related regulators in UVM and suggest potential novel targets for the diagnosis, prognosis and therapy of UVM.
Similar content being viewed by others


Introduction
Uveal melanoma originates from melanocytes in the eye with approximately 85% of tumor cases arising from choroid [1], it can also occur in the ciliary body or iris, and predominantly affects adults relative to children [2]. Up to 45% of uveal melanoma patients have metastasis within 15 years, that is prone to metastasize to liver (around 70%-90%) and with poor prognosis. Lack of effective treatment options on metastatic cases result in short survival time, commonly within one year [3, 4]. In recent years, numerous studies regarding the treatment of patients with metastatic UVM were focused on immunotherapy, such as immune checkpoint inhibitors [5, 6]. However, recent studies have found that certain inhibitors, such as anti-CTLA-4 antibody lpilimumab, have not achieved satisfactory results in the treatment of UVM patients [7]. Thus, the search for better biomarkers is extremely urgent, which are of profound importance in assessing patients' clinical outcomes and long-term prognosis. Epigenetic regulations, including lysine methylation and RNA modification, ubiquitously affect gene functions through the post-transcriptional modification, and these processes are vital to tumorigenesis [8]. It can potentially be applied as biomarker and of therapeutic targets for carcinomatosis [9]. RNA modification in epigenetics, especially RNA methylation, accumulating publications have been conducted on the topic in the last decade, excess of 100 types RNA modifications have been identified, the modification of m7G is one of the most universal forms of those, along with N6-methyladenosine (m6A) [10, 11]. At present, various lines of evidence show that the dysregulation of tRNA and its corresponding modification catalytic enzymes are involved in a variety of human cancers [12]. The most highly modified RNA molecule in cells is transfer RNA (tRNA) [13], which function as adapter molecules at post-transcription stage. The modification of m7G primarily occurs on tRNA within variable region located at position 46, produced by Methyltransferase for tRNA (m7G46) [14]. Methyltransferase methyltransferase-like protein-1(METTL1) and its’ partner WD repeat Domain 4 (WDR4), as catalytic enzymes of m7G methylation, that play a vital role in m7G methylation m7G [15]. Over the past few years, the role of m7G in cancer progression has been revealed in several studies [15,16,17,18], but the function and molecular mechanism of m7G in UVM is far from clear.
In present study, we collected data from the TCGA and GEO databases that including the clinical, mutation annotations and mRNA expression information of UVM patients. We constructed m7G-related risk model composed of four genes based on TCGA cohort of 80 UVM patients and further validated by two GEO cohort. Moreover, we analyzed the association with risk score based on the risk model and immune status in UVM patients. Our study provides a novel perspective on clinical diagnosis, prognosis and therapy of UVM patients.
Materials and methods
Dataset selection
The clinical, annotations of mutation and mRNA expression data of 80 UVM patients were collected from TCGA (https://portal.gdc.cancer.gov) as a training set of the m7G-related risk model. The clinical and mRNA expression data of GSE22138 (N = 63) and GSE84976 (N = 28) were selected from GEO (https://www.ncbi.nlm.nih.gov/geo), as the validation set by integration [19, 20]. Abstracts providing information about each dataset are presented in Table 1. We eliminated batch effects using the R packages "sva" and "limma". In the case of the same gene was probed with multiple probes, the expression of gene was measured by taking its average value. Using published reviews and Gene Set Enrichment Analysis (GSEA, http://www.gsea-msigdb.org) searches for "7-methylguanosine", 29 genes related to m7G were identified [14, 21, 22](Supplemental Table 1).
Development and validation of the prognostic risk model
Genes of m7G-related regulators prominently associated with prognosis of OS were sifted by Kaplan–Meier and Cox regression of univariate (p < 0.05). Following that, regression analysis on the basis of the least absolute shrinkage and selection operator (LASSO), a popular penalization method, was utilized to process the prognostic genes obtained by applying the R package "glmnet" [23]. After the establishment of the prognostic risk model by LASSO, the result of the ten-fold cross-validation was used as the regularization parameter lambda (λ) of the risk model. We used the following calculation equation to measure the risk score of each sample: \(Riskscore={\sum }_{i=1}^{i}\left(\text{Coefi}*\text{ExpGenei}\right)\).
ExpGene stood for the expression of each m7G-related gene, and coef stood for the coefficient of Cox regression for each gene and n as the number of genes included in the formula. Patients of UVM were separated into two cohorts, including high risk cohorts and low risk cohorts, based on their mid-value. ROC curves based on time-dependent were implemented using the R package "survminer" and "timeROC" to evaluate the precision of prediction [24]. For the purpose of comparing prognostic diversities between high-risk and low-risk cohorts, we executed Kaplan–Meier curves using the R package "survminer". R package "ggplot2" was used for Principal Component Analysis (PCA) analysis. In order to test if the risk model was a reliable independent prognostic factor based on OS, we enforced the analysis of the association with clinical characteristics and risk score by Cox regression, both univariate and multivariate.
Comparison of cohorts at high and low risk on the basis of the m7G-related risk model
The DEGs were calculated by comparing cohorts at high and low risk. Subsequently, |log (FC)|> 1 and adjustments-p-value < 0.05 were the filter conditions to screen out prominent Differently Expressed Genes (DEGs). The analyses of Kyoto Encyclopedia of Genes and Genomes (KEGG, https://www.kegg.jp) and Gene Ontology (GO, http://geneontology.org) were implemented by R package "clusterProfiler". Immune-related pathways score and immune cells score were calculated for ssGSEA (Single Sample GSEA) by R package "GSEABase" and "GSVA". The proportion of immune matrix components in the tumor microenvironment (TME) was calculated by R package "estimate" to generate the immune score, matrix score, and Estimate score [25]. Thereafter, we calculated immune, matrix, and estimation scores for high-risk and low-risk cohorts, respectively.
Statistical analyses
These analyses of statistics were enforced by the R software (R Foundation for Statistical Computing, 64-bit, R 4.1.2). Programming language Perl was used for bulk data processing (Perl 5.32.1). Statistical analyses of correlation and difference were carried out using R package "limma". Prominent differences were measured by the Wilcoxon rank-sum test in variables that were continuous between the two cohorts. The R packages "survminer" and "survival" were applied to implement the analysis of Kaplan–Meier. Statistical significance is considered at p-value < 0.05.
Results
Research process and clinical characteristics
Eighty UVM samples from TCGA database were selected as the training set, and 91 samples of GSE22138 and GSE84976 obtained from GEO database were chosen for validation set after data integration with R package "sva" to remove batch effect. A summary of clinical parameters is provided in Table 2. Next, using the DNA copy number variation (CNV) frequency data of UVM, we analyzed and visualized 29 m7G-related genes (Fig. 1a). The frequency of gain of AGO2 and NUDT3 was significantly higher than its loss, while the results of EIF4E3, EIF4G3, NCBP2 and NUDT16 were contrary. The positions of 29 m7G-related genes on different chromosomes were illustrated in Fig. 1b. Notably, among the first three genes in which the frequency of loss was significantly greater than gain, they were located on chromosome 3.

DNA copy number variation (CNV) frequency data of 29 genes associated with m7G and the corresponding positions of chromosomes. a Gene CNV frequency data of 29 m7g-related genes with UVM, gain is represented by the red dot and loss is represented by green dot. b The positions of 29 m7G-related genes on different chromosomes
The prognostic risk model is developed and validated on the basis of m7G-related regulators
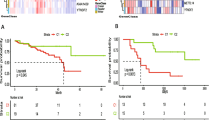
Following extracting the expression data of 29 m7G-related genes from 80 UVM samples from TCGA, we carried out Cox regression of Kaplan–Meier and univariate analyses. Nine m7G-related genes were identified, and they were remarkably associated with prognosis in terms of OS. Integrating the nine genes into Cox regression of LASSO, the LASSO coefficient curve consisting of four m7G-related prognostic genes was obtained, with the minimum parameter serve as the penalty regularization parameter lambda (λmin = 0.0411, Fig. 2a-b and Supplemental Table 1–3). The LASSO Cox regression model is: Risk score = (1.170190103 × mRNA expression of EIF4E2) + (-0.470940387 × mRNA expression of NUDT16) + (0.55003035 × mRNA expression of SNUPN) + (0.945486337 × mRNA expression of WDR4) (Table 3). Using the mid-value of risk score as the cut-off, UVM patients were categorized into high and low risk cohorts.

Developing and validating the prognostic risk model on the basis of m7G-related regulators. a LASSO coefficient profiles of the four genes which belong to the m7G-related risk model. bThe penalty regularization parameter lambda (λ) was determined by the result of the ten-fold cross-validation. c, d The curves of Kaplan–Meier analysis for TCGA cohort (c) and GEO cohort (d). e, f ROC analysis for TCGA cohort (e) and GEO cohort (f)
According to the Kaplan–Meier curve (p < 0.001), the cohort at high risk in Fig. 2c exhibited poor performance. Subsequently, the risk model’s predictive ability the OS prognosis by calculating 1, 3 and 5-years AUC values (AUC at 1, 3 and 5-years was 0.770, 0.856, and 0.869, respectively, as shown in Fig. 2e). The risk model demonstrated reliable long-term prognostication. Consistent with the training set, the Kaplan–Meier curve for the validation set showed poor prognosis in the high risk cohort (p < 0.001, Fig. 2d), Additionally, Fig. 2f was generated through ROC analysis, displaying AUC value at 1, 3 and 5-years (0.632, 0.808 and 0.828, respectively). These findings further support the predictability of the risk model for the prognosis of OS.
To comprehensively assess the risk model, we calculated and visualized both the risk plot and PCA distribution for the training and validation sets, respectively (Fig. 3a-d). UVM Patients from the cohort at high risk exhibited a poor prognosis compared to those from low risk cohort. Furthermore, PCA analysis illustrated that the risk model effectively distinguish between the high and low risk cohorts. Therefore, the risk model, based on four genes related to m7G-related regulators was successfully established to assess the prognosis of UVM patients.

The distribution of the risk plot and PCA. a, b Analyses of the distribution based on risk score and survival status for TCGA cohort (a) and GEO cohort (b). c, d PCA plot of TCGA cohort (c) and GEO cohort(d)
Correlation between clinical characteristics and risk score
To rigorously evaluate the risk model as robust independent indicator of OS prognosis in UVM patients, both univariate and multivariate Cox regression analyses were enforced between the high-risk and the low-risk cohorts. We integrated clinical information from 80 UVM samples in TCGA, including age, gender, stage, tumor diameter, tumor thickness and extrascleral extension. Univariate Cox regression results revealed that age, stage, tumor diameter, extrascleral extension and risk score were prominently independent variables of OS prognosis in UVM patients (p < 0.05, Fig. 4a). In multivariate Cox regression analysis, risk score remained an independent variable of OS prognosis in UVM patients (p < 0.05, Fig. 4b). This underscores the validity of the risk model based on m7G-related regulators in predicting UVM prognosis. Additionally, we performed a ROC analysis based on time-dependent for both risk score and clinical characteristics. As depicted in Fig. 4c, the AUC of risk score was 0.869, surpassing the AUC that based on clinical characteristics. This result strongly indicates that risk score provide a more accurate prediction in prognosis compared to clinical characteristics.

Correlation between clinical characteristics and risk score. The Cox regression of univariate (a) and multivariate (b) between clinical characteristics and risk score. c ROC analysis on risk score and clinical characteristics
Gene set enrichment analysis
To further elucidate differences in pathway and gene function between high-risk and low-risk cohorts categorized based on risk model, we identified the DEGs of TCGA cohort using condition (p < 0.05 and |logFC|> 1). Subsequently, we performed KEGG and GO based on these DEGs. In KEGG pathway enrichment analysis, the result revealed that the risk model was closely correlated with phagosome, infection and cell adhesion molecules (Fig. 5a). In GO function enrichment analysis, T cell activation exhibits the most significant enrichment, followed by leukocyte-mediated immunity and lymphocyte mediated immunity, among others. The risk model based on m7G-related regulators exhibited a strong association with T cell function and immune (Fig. 5b). These findings suggest that the identified risk model might play a crucial role in modulating T cell activities and immune responses.

KEGG and GO enrichment analysis. The bubble belongs to enrichment analyses of KEGG (a) and GO (b) in DEGs for TCGA cohort
Correlation between somatic mutations and risk score
In the high and low risk cohorts, R package "maftools" was employed to analyze the distribution of mutational differences between m7G-related genes. The top twenty genes with the highest mutation rates were selected and visualized in Fig. 6a-b. The waterfall plots depicted that the most significant pre-mutation gene in low risk cohort, was GNA11, while, in the low risk cohort was GNAQ. These findings suggest distinct mutational landscapes in m7G-related genes between the two risk groups.

Mutation map of cohorts at high and low risk cohorts. Mutation map of the top twenty mutated genes for (a) high-risk cohort and (b) low-risk cohort
Correlation between immune characteristics and risk score
The risk model we developed may be closely related to immunity in gene set enrichment analysis. Therefore, we analyzed the differences in immune characteristics between high-risk and low-risk cohorts from the TCGA-UVM. In our investigation using ssGSEA, we have found that most immune cell subpopulations and functions were significantly enhanced in the high-risk cohort (p < 0.05, Fig. 7a,b), it suggests that the UVM high-risk cohort exhibits over expression of immune-related functions. Next, we compared the mRNA expression of genes related to immune checkpoints between the two cohorts. Immune checkpoint-related genes exhibited higher expression levels in high-risk cohorts, as depicted in (Fig. 7c). Using R package "estimate" analysis to reflect the immune microenvironment landscape of tumor, we found that immune, stromal and estimate scores of high-risk cohort were significantly enhanced (p < 0.001), each correlation coefficient was greater than 0.34 in absolute value (Fig. 7d). In addition to immune microenvironment, tumor stemness scores, encompassing DNA methylation profiles and RNA stemness scores, also played a pivotal role in tumor progression. Consistent with the results of immune microenvironment, DNA methylation profiles showed a strikingly positive relationship with UVM risk score (R = 0.49, p < 0.05, Fig. 7e), while RNA stemness score did not exhibit a remarkable significant positive correlation in our results (R = 0.2, p = 0.074, Fig. 7f). These findings provide further support for our earlier findings that risk score based on m7G-related risk models is closely related to immune activation.

Correlation between immune characteristics and risk score. The immune cell subpopulations (a) and functions (b) between cohorts at high and low risk based on the ssGSEA. c The mRNA expression of each gene related to immune checkpoints of cohorts at high and low risk. d The violin figure of immune, stromal and ESTIMATE scores for cohorts at high and low risk. e DNAss. f RNAss
Discussion
M7G, N7-methylguanosine, represents a one form of RNA modification. This modification is known to enhance mRNA stability and occurs prior to changes at the protein level, suggesting its potential advantage in predicting disease progression. Focusing solely on modifications to individual genes may limit the accuracy and comprehensiveness needed to predict the course of a disease. In this study, nine m7G-related prognostic regulators were identified by Kaplan–Meier and univariate Cox analyses in the TCGA cohort. The risk model for UVM prognosis was established using LASSO Cox regression, which consisted of four m7G-related genes (EIF4E2, NUDT16, SNUPN and WDR4). To evaluate the efficacy of m7G-related risk model, we stratified the TCGA cohort was into high-risk and low-risk cohorts with the mid-value approach. We then carried out Kaplan–Meier curves and ROC analysis between these two cohorts. Strikingly statistical differences show high-risk cohort exhibited worse prognosis compared to those in low-risk counterparts. The AUC at 1, 3 and 5-years were 0.770, 0.856, and 0.869, respectively, suggesting strong predictive capability on prognosis of OS of UVM. Furthermore, we conducted both univariate and multivariate Cox regression to assess the correlation between risk score and major clinical characteristics. Our results confirmed that our four genes m7G-related risk model was reliably independent of other variables that predicted UVM outcomes. To validate our findings, we applied in GEO database and visualized the risk plot and PCA distributions. the AUC at 1, 3 and 5-years was 0.632, 0.808 and 0.828, respectively. This validation further demonstrated that the m7G-related risk model was a reliably independent indicator of OS prognosis.
Among the four prognostic genes of risk model, WDR4 plays a non-catalytic role in m7G methyltransferase complex. The complex formed by METTL1 and WDR4 catalyzes the methylation at position 46 of tRNA, that primarily stabilizing WDR4 structure [26, 27]. It had been demonstrated experimentally that METTL1/WDR4 can regulate the development of cancer through tRNA modification of m7G methylation in intrahepatic cholangiocarcinoma, lung cancer and nasopharyngeal carcinoma [15,16,17,18]. Jieyi Ma et al. observed a striking deceleration in lung cancer progression by knocking down METTL1/WDR4 [17]. Eukaryotic translation initiation factor 4E family member 2 (EIF4E2), belongs to eukaryotic promoter factor, is a cap-binding protein that binds to 7-methylguanosine-containing mRNA caps during translation initiation [28]. Nudix hydrolase 16 (NUDT16) is a nuclear RNA decapping protein, Taylor MJ et al. reported that NUDT16 is capable of binding and hydrolyzing the m7G cap from the 5'-end of RNA in vitro analysis of the insect ortholog [29]. Another study of Shaohong Peng et al. found that NUDT16 silencing leas to apoptosis and DNA damage [30]. Snurportin 1 (SNUPN), an adaptor protein, is a nuclear import receptor specifically associated with spliceosomal snRNPs U1, U2, U4 and U5. Moreover, SNUPN encoded proteins interact specifically with m3G-Cap and m7G-Cap [31]. In addition to NUDT16 being a protective factor, the other three genes (WDR4, EIF4E2, and SNUPN) are risk factors that accelerate tumor progression. Our results regarding m7G modification in the above four genes were consistent with the previous studies that have been reported to affected tumor progression.
Multiple lines of evidence have demonstrated RNA methylation closely correlated with tumor immunity, highlighting its crucial clinical implications. In our study, KEGG pathway enrichment analysis revealed that the main pathways affected are phagosome, infection and cell adhesion molecules, Meanwhile, the primary outcomes of GO functional enrichment analysis included T cell activation top the BP section, leukocyte-mediated immunity and lymphocyte-mediated immunity, indicating a close association between immune response and the risk score calculated by the m7G-related risk model. Subsequently, the ssGSEA and TME analyses were carried out and both indicating a higher level of immunoreaction in the high-risk cohort. It’s well- known that to prevent damage to non-regenerative tissues such as neural retina by immunoreactivity, normal organism has the immune privilege to their eyeballs [32]. The Kaplan–Meier curve of Fig. 3c, we observed strikingly poor prognosis in the high-risk cohort. Combined with the previous results, we hypothesize that the dysregulation of m7G related genes in UVM patients is closely related to the enhancement of ocular immunoreaction, especially T cells and leukocytes. This hyperactive immune response compromises the eyeball's local immunity, contributing to worse outcomes in UVM patients. based, The comprehensive AUC value based on our 4 genes risk model of m7G-related regulators is 0.869, precisely predicting the prognosis of UVM patients In contrast to the significant strides made in cutaneous melanoma treatment the impact of immune checkpoint inhibitors on UVM is constrained, with the evidence of drug resistance [33]. UVM patients exhibiting high scores of m7G-related risk model displayed increasing abundant of immune infiltration, notably characterized by a general up-regulation of immune checkpoint related genes. This suggested that UVM patients with high score exhibit higher sensitivity to immune checkpoint inhibitors. Clinical application of the m7G-related risk model can help doctors timely identifying UVM patients suitable for immunotherapy. In conclusion, UVM patients exhibiting high risk score stratified by the m7G-related risk model are more likely to respond positively to immunotherapy, which has strikingly implication for application of immune checkpoint inhibitors in clinical settings. However, it's crucial to acknowledge the limitations in our research such as limited samples incorporated. This model may necessitate more prospective data from multiple center studies.
Conclusions
In this study, we utilized data from TCGA and GEO database to formulate a prognostic risk model comprising four m7G-related genes, suggesting that m7G based modification may shed a novel perspective on diagnosis, prognosis, survival and therapeutic targets correlated with UVM.
Availability of data and materials
The original data involved in this work comes from the website: https://portal.gdc.cancer.gov; https://www.ncbi.nlm.nih.gov/geo; http://www.gsea-msigdb.org. The code and results used to support the findings of this study are available from the corresponding author.
Abbreviations
- UVM:
-
Uveal melanoma
- m7G:
-
N7-methylguanosine
- TCGA:
-
The Cancer Genome Atlas
- GEO:
-
Gene Expression Omnibus
- ROC:
-
Receiver Operating Characteristic
- AUC:
-
Area Under the Curve
- m6A:
-
N6-methyladenosine
- METTL1:
-
Methyltransferase methyltransferase-like protein-1
- WDR4:
-
WD repeat Domain 4
- GSEA:
-
Gene Set Enrichment Analysis
- LASSO:
-
Least absolute shrinkage and selection operator
- PCA:
-
Principal Component Analysis
- DEGs:
-
Differently Expressed Genes
- TME:
-
Tumor microenvironment
- ssGSEA:
-
Single Sample GSEA
- CNV:
-
DNA copy number variation
- EIF4E2:
-
Eukaryotic translation initiation factor 4E family member 2
References
McLaughlin CC, Wu XC, Jemal A, et al. Incidence of noncutaneous melanomas in the U.S. Cancer. 2005;103(5):1000–7.
Chattopadhyay C, Kim DW, Gombos DS, et al. Uveal melanoma: from diagnosis to treatment and the science in between. Cancer. 2016;122(15):2299–312.
Ponti A, Denys A, Digklia A, et al. First-line selective internal radiation therapy in patients with uveal melanoma metastatic to the liver. J Nucl Med. 2020;61(3):350–6.
Ortega MA, Fraile-Martínez O, García-Honduvilla N, et al. Update on uveal melanoma: translational research from biology to clinical practice. Int J Oncol. 2020;57(6):1262–79.
Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(1):23–34.
Weber JS, D’Angelo SP, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16(4):375–84.
Rozeman EA, Prevoo W, Meier MA, et al. Phase Ib/II trial testing combined radiofrequency ablation and ipilimumab in uveal melanoma (SECIRA-UM). Melanoma Res. 2020;30(3):252–60.
Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150(1):12–27.
Hogg SJ, Beavis PA, Dawson MA, et al. Targeting the epigenetic regulation of antitumour immunity. Nat Rev Drug Discovery. 2020;19(11):776–800.
Motorin Y, Helm M. RNA nucleotide methylation: 2021 update. Wiley Interdisciplinary Reviews: RNA. 2022;13(1):e1691.
Barbieri I, Kouzarides T. Role of RNA modifications in cancer. Nat Rev Cancer. 2020;20(6):303–22.
Suzuki T. The expanding world of tRNA modifications and their disease relevance. Nat Rev Mol Cell Biol. 2021;22(6):375–92.
Li J, Zhu WY, Yang WQ, et al. The occurrence order and cross-talk of different tRNA modifications. Sci China Life Sci. 2021;64(9):1423–36.
Tomikawa C. 7-Methylguanosine modifications in transfer RNA (tRNA). Int J Mol Sci. 2018;19(12):4080.
Chen B, Jiang W, Huang Y, et al. N7-methylguanosine tRNA modification promotes tumorigenesis and chemoresistance through WNT/β-catenin pathway in nasopharyngeal carcinoma. Oncogene. 2022;41(15):2239–53.
Dai Z, Liu H, Liao J, et al. N7-Methylguanosine tRNA modification enhances oncogenic mRNA translation and promotes intrahepatic cholangiocarcinoma progression. Mol Cell. 2021;81(16):3339–55.
Ma J, Han H, Huang Y, et al. METTL1/WDR4-mediated m7G tRNA modifications and m7G codon usage promote mRNA translation and lung cancer progression. Mol Ther. 2021;29(12):3422–35.
Orellana EA, Liu Q, Yankova E, et al. METTL1-mediated m7G modification of Arg-TCT tRNA drives oncogenic transformation. Mol Cell. 2021;81(16):3323-3338. e14.
Laurent C, Valet F, Planque N, et al. High PTP4A3 phosphatase expression correlates with metastatic risk in uveal melanoma patients. Can Res. 2011;71(3):666–74.
van Essen TH, van Pelt SI, Bronkhorst IH, et al. Upregulation of HLA expression in primary uveal melanoma by infiltrating leukocytes. PLoS ONE. 2016;11(10):e0164292.
Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci. 2005;102(43):15545–50.
Mootha VK, Lindgren CM, Eriksson KF, et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267–73.
Wang H, Lengerich BJ, Aragam B, et al. Precision Lasso: accounting for correlations and linear dependencies in high-dimensional genomic data. Bioinformatics. 2019;35(7):1181–7.
Heagerty PJ, Lumley T, Pepe MS. Time‐dependent ROC curves for censored survival data and a diagnostic marker. Biometrics. 2000;56(2):337–44.
Yoshihara K, Shahmoradgoli M, Martínez E, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4(1):1–11.
Alexandrov A, Martzen MR, Phizicky EM. Two proteins that form a complex are required for 7-methylguanosine modification of yeast tRNA. RNA. 2002;8(10):1253–66.
Alexandrov A, Grayhack EJ, Phizicky EM. tRNA m7G methyltransferase Trm8p/Trm82p: evidence linking activity to a growth phenotype and implicating Trm82p in maintaining levels of active Trm8p. RNA. 2005;11(5):821–30.
Rosettani P, Knapp S, Vismara M-G, et al. Structures of the human eIF4E homologous protein, h4EHP, in its m7GTP-bound and unliganded forms. J Mol Biol. 2007;368(3):691–705.
Taylor MJ, Peculis BA. Evolutionary conservation supports ancient origin for Nudt16, a nuclear-localized, RNA-binding, RNA-decapping enzyme. Nucleic Acids Res. 2008;36(18):6021–34.
Peng S, Guo P, Lin X, et al. CAG RNAs induce DNA damage and apoptosis by silencing NUDT16 expression in polyglutamine degeneration. Proc Natl Acad Sci. 2021;118(19):e2022940118.
Huber J, Cronshagen U, Kadokura M, et al. Snurportin1, an m3G-cap-specific nuclear import receptor with a novel domain structure. EMBO J. 1998;17(14):4114–26.
Keino H, Horie S, Sugita S. Immune privilege and eye-derived T-regulatory cells. J Immunol Res. 2018;2018:1679197.
Masaoutis C, Kokkali S, Theocharis S. Immunotherapy in uveal melanoma: Novel strategies and opportunities for personalized treatment. Expert Opin Investig Drugs. 2021;30(5):555–69.
Acknowledgements
Not applicable.
Funding
This work was sponsored by Shanghai Shenkang Three Years Program (Grant number SHDC2020CR2054B).
Author information
Authors and Affiliations
Contributions
Conception and design: PW and QZ; Data curation and methodology: PW, QZ, and PZ; Analysis and interpretation of data: QZ, LC, and QL; Writing of the manuscript: PW; Review of the manuscript: CJ; Study supervision: PW and LC. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study is a public data based further analysis and therefore does not require ethical approval.
Consent for publication
Not applicable.
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wu, P., Zhang, Q., Zhong, P. et al. Development of a prognostic risk model of uveal melanoma based on N7-methylguanosine-related regulators. Hereditas 161, 22 (2024). https://doi.org/10.1186/s41065-024-00324-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41065-024-00324-0