
Abstract
Background
Suanzaoren Decoction (SZRD), a well-known formula from traditional Chinese medicine, has been shown to have reasonable cognitive effects while relaxing and alleviating insomnia. Several studies have demonstrated significant therapeutic effects of SZRD on diabetes and Alzheimer’s disease (AD). However, the active ingredients and probable processes of SZRD in treating Alzheimer’s with diabetes are unknown. This study aims to preliminarily elucidate the potential mechanisms and potential active ingredients of SZRD in the treatment of Alzheimer’s with diabetes.
Methods
The main components and corresponding protein targets of SZRD were searched on the TCMSP database. Differential gene expression analysis for diabetes and Alzheimer’s disease was conducted using the Gene Expression Omnibus database, with supplementation from OMIM and genecards databases for differentially expressed genes. The drug-compound-target-disease network was constructed using Cytoscape 3.8.0. Disease and SZRD targets were imported into the STRING database to construct a protein-protein interaction network. Further, Gene Ontology and Kyoto Encyclopedia of Genes and Genomes analyses were performed on the intersection of genes. Molecular docking and molecular dynamics simulations were conducted on the Hub gene and active compounds. Gene Set Enrichment Analysis was performed to further analyze key genes.
Results
Through the Gene Expression Omnibus database, we obtained 1977 diabetes related genes and 622 AD related genes. Among drugs, diabetes and AD, 97 genes were identified. The drug-compound-target-disease network revealed that quercetin, kaempferol, licochalcone a, isorhamnetin, formononetin, and naringenin may be the core components exerting effects. PPI network analysis identified hub genes such as IL6, TNF, IL1B, CXCL8, IL10, CCL2, ICAM1, STAT3, and IL4. Gene Ontology and Kyoto Encyclopedia of Genes and Genomes analyses showed that SZRD in the treatment of Alzheimer’s with diabetes is mainly involved in biological processes such as response to drug, aging, response to xenobiotic, and enzyme binding; as well as signaling pathways such as Pathways in cancer, Chemical carcinogenesis - receptor activation, and Fluid shear stress and atherosclerosis. Molecular docking results showed that licochalcone a, isorhamnetin, kaempferol, quercetin, and formononetin have high affinity with CXCL8, IL1B, and CCL2. Molecular dynamics simulations also confirmed a strong interaction between CXCL8 and licochalcone a, isorhamnetin, and kaempferol. Gene Set Enrichment Analysis revealed that CXCL8, IL1B, and CCL2 have significant potential in diabetes.
Conclusion
This study provides, for the first time, insights into the active ingredients and potential molecular mechanisms of SZRD in the treatment of Alzheimer’s with diabetes, laying a theoretical foundation for future basic research.
Graphical Abstract

Highlights
• SZRD may improve Alzheimer’s with diabetes through potential active ingredients and hub genes.
• licochalcone a, isorhamnetin, kaempferol, quercetin, and formononetin are potential active ingredients of SZRD for the treatment of Alzheimer’s with diabetes.
• IL6, TNF, IL1B, CXCL8, IL10, CCL2, ICAM1, STAT3 and IL4 are hub genes and have a strong binding capacity to potential active ingredients.
Similar content being viewed by others


Introduction
Diabetes is increasingly prevalent globally, with the highest incidence observed in the age group of 75–79 years old [1]. In 2021, global healthcare expenditures associated with diabetes were estimated at $966 billion, with a projected increase to $10.54 trillion by 2045 [2]. AD is the leading neurodegenerative cause of dementia, responsible for 50–70% of cases of neurodegenerative dementia. The estimated global prevalence of dementia is around 44 million people, and this number is projected to triple by 2050 [3]. Diabetes can result in brain tissue damage and cognitive dysfunction, and it is also recognized as a risk factor for AD [4, 5]. Epidemiological studies have confirmed that individuals with diabetes have an elevated risk of dementia compared to those without diabetes [6]. Studies have shown that the risk of dementia in patients with diabetes is associated with the prevalence of mild cognitive dysfunction progressing to dementia [7]. Cognitive dysfunction in individuals with diabetes plays a significant role in the relationship between diabetes and AD. Hence, the discovery of novel therapeutic drugs for managing Alzheimer’s with diabetes holds profound implications for individuals with both diabetes and AD.
Traditional Chinese medicine has garnered growing attention as a potential treatment for cognitive impairments associated with diabetes. SZRD is a Chinese herbal formula that originated from the book “Jin Kui Yao Lue” written by Zhang Zhongjing in the Han Dynasty, and it comprises five medicinal herbs, namely Ziziphus jujuba Mill (Suanzaoren, SZR), Glycyrrhiza uralensis Fisch (Gancao, GC), Anemarrhena asphodeloides Bunge (Zhimu, ZM), Poria cocos (Schw.) Wolf. (Fuling, FL), and Ligusticum acuminatum Franch. (Chuanxiong, CX), known for their calming, nourishing, and insomnia-treating effects. Despite being a traditional herbal prescription for insomnia, SZRD has also been utilized in the treatment of other ailments. Studies have demonstrated that SZRD can elevate the levels of neurotransmitters, including 5-HT, DA, and GABA, in the brain, potentially contributing to its effectiveness in treating insomnia [8]. Moreover, SZRD has exhibited the ability to enhance learning and memory in mouse models of AD [9]. In addition, Glycyrrhiza uralensis Fisch [10] and Anemarrhena asphodeloides Bunge [11] have been found to improve diabetes. We hypothesize that SZRD holds promising potential for the treatment of cognitive impairments and diabetes. Further investigation and elucidation of its underlying compounds and targets are warranted to comprehensively understand its therapeutic effects in cognitive disorders.
The network pharmacology approach was originally introduced as a novel avenue for identifying new drug candidates and repurposing existing ones from intricate network models [12]. The study of herbal medicines in disease treatment poses unique challenges due to their complex composition, multiple targets, and broad therapeutic signaling pathways [13]. Hence, utilizing network pharmacology to elucidate the effects of traditional herbal formulations on complex diseases and predict potential compounds and targets represents a crucial approach. Molecular docking, as a structure-based and computer-assisted drug design method, holds a pivotal role in drug discovery and research [14]. Molecular dynamics simulation, capable of capturing the real-time trajectory of macromolecules [15], has found widespread use in various fields, including biology. We have employed a multi-faceted approach encompassing bioinformatics, network pharmacology, molecular docking, and molecular dynamics simulation to precisely target herbal medicines for disease treatment.
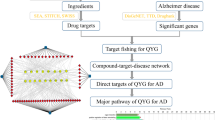
SZRD has demonstrated promising efficacy in improving cognitive dysfunction, and some studies have also reported its potential in lowering blood glucose levels. We posit that SZRD may hold promise as an herbal medicine for treating Alzheimer’s with diabetes, albeit challenges persist in identifying its composition and targets. Therefore, this study has integrated bioinformatics, network pharmacology, and molecular docking to predict the active compounds, potential targets, and underlying molecular mechanisms of SZRD in Alzheimer’s with diabetes, aiming to contribute to the field of traditional Chinese medicine and provide a reference and foundation for future researchers. The experimental workflow is illustrated in Fig. 1.

Workflow of the network pharmacological investigation strategy of SZRD in the treatment of diabetes and AD. Four parts include target preparation, Hub gene, and network analysis, GO and KEGG analysis, molecular docking verification, and GSEA analysis
Methods
Component target prediction
Traditional Chinese Medicine Pharmacology Systematic Pharmacology (TCMSP, https://old.tcmsp-e.com/tcmsp.php) database based on the systematic pharmacology of Chinese medicine [16], It contains a rich variety of herbs, chemical components, and 12 important properties related to ADME required for drug screening and evaluation. Oral bioavailability (OB) is the rate and extent to which a drug is absorbed into the circulation. Drug similarity (DL) is the similarity between the component and the marketed drug [17]. Based on the OB principle, we used OB ≥ 30% and DL ≥ 0.18 in the TCMSP component screening [18], while reviewing the literature for further collection. Uniport (http://www.uniprot.org/) was used to transform the obtained targets in terms of gene symbols. Admetlab 2.0 (https://admetmesh.scbdd.com/) is an online pharmacokinetic and toxicity prediction program [19]. It was used to predict drug absorption, distribution, metabolism, excretion, and toxicity profiles. The SMILES of the compounds are entered into Admetlab 2.0 to obtain the absorption, distribution, and toxicity profiles of the components, etc.
Disease target prediction
The Gene Expression Omnibus Dataset (GEO, http://www.ncbi.nlm.nih.gov/geo/) database is a website for disease gene expression, and we used “diabetes” and " Alzheimer’s disease " as search strategies, limiting species to “homo spines”. We selected datasets for diabetes (GSE15653) and AD (GSE132903), using “limma”, “pheatmap”, “ggplot2 “, “ggsci”, “dplyr”, “org.Hs.eg.db” and “patchwork” packages were used to visualize the differentially expressed genes(DEG), volcanoes and heatmaps, and the threshold values for DEG identification were |logfc|>0.5 and P < 0.05 [20]. The search for AD and diabetes-related targets was performed by Genecards (https://www.genecards.org/) and OMIM (https://www.omim.org/) using the keywords “AD” and “diabetes”. The Relevance score was restricted to the second average in Genecards to accurately screen the relevant targets, and all the targets of diabetes and AD were combined and removed the duplicate values to draw a Venn diagram.
Construction of drug-compound-target-disease (D-C-T-D) networks and protein-protein interaction (PPI) networks
To better analyze the connection between drugs, components, and targets, we built a “D-C-T-D” network in Cytoscape 3.8.0 software, Cytoscape can calculate the parameters of each node in the network graph, such as degree, betweenness centrality (BC), closeness centrality (CC), etc. [21, 22]. All these parameters allow an in-depth analysis of the properties of the nodes in the interaction network, and we used the degree, BC, and CC to filter the top-ranked components and targets, regarded as playing a central role. The STRING network was constructed (https://string-db.org/) as a database for analyzing the relationships between proteins [23]. Using this database, we construct a “PPI” network that captures the interactions between intersecting targets and collects targets with strong connections. The scoring condition was set to > 0.70 and the selected target proteins were restricted to Homo sapiens. In the PPI network, edges represent protein-protein associations, and the more lines there are, the greater the correlation. Screening of key targets and analysis followed.
Hub gene extraction and MCODE analysis
PPI networks consist of nodes, edges, and connecting lines, and it is generally believed that the most critical nodes are hub genes. Cytohubba (http://apps.cytoscape.org/apps/cytohubba) is a new Cytoscape plugin for ranking and extracting biological networks based on various network features for central or potential target elements based on various network features. Cytohubba has 11 methods to study networks from different perspectives, of which maximum population centrality (MCC) is the best one [24]. We used the MCC of Cytohubba to identify the top 10 hub genes from PPI networks. Metascape (https://metascape.org/gp/index.html) is an efficient tool in the era of big data, combining functional enrichment, interactome analysis, gene annotation, and member search to provide a comprehensive gene list annotation and analysis resource [25]. We entered all intersecting genes into Metascape for MCODE analysis.
GO and KEGG pathway enrichment analysis
R packages such as “org.Hs.eg.db”, “clusterprofiler”, “enrichplot”, “ggplot2”, “ggnewscale” and “DOSE” were used for GO and KEGG enrichment analysis and result plotting. GO is divided into three categories, namely biological process (BP), Cellular component (CC), and Molecular function (MF), and KEGG enrichment analysis is a way to analyze the pathway enrichment of genes. All intersecting targets were analyzed and the top ten biological processes of BP, CC, MF and KEGG in GO were selected for graphical visualization.
Molecular docking
Molecular docking is one of the most commonly used methods for structure-based drug design [26]. Molecular docking of active compounds and hub genes was performed using autodocktools-1.5.7 software. First, the 3D structure of the active compound was downloaded from PubChem (http://Pubchem.ncbi.nlm.nih.gov/); then, the water was removed, and hydrogen atoms were added and converted to PDBQT format using autodocktools. Download the PDB format of the relevant target at RSCB-PDB (https://www.rcsb.org/) and remove the ligands and water molecules using PYMOL software. Then, import them into autodocktools, add hydrogen atoms, calculate the charges, convert them to PDBQT format and perform molecular flexible docking, selecting the docking model with the lowest binding energy among them. Finally, the 3D docking results were visualized using PYMOL software and the 2D docking results were visualized using Discovery Studio software 2017. Among them, a docking score AFFINITY <-4.25 KCAL/MOL−1 considered a binding activity between ligand and target, a score <-5.0 KCAL/MOL−1 indicated a better binding activity, and a score <-7.0 KCAL/MOL−1 a strong docking activity between the two [27].
Molecular dynamics simulation
All-atom molecular dynamics simulations were performed separately based on the small molecule and protein complexes obtained from the above docking as initial structures, and the simulations were performed using AMBER 18 software [28]. Before the simulations, the charges of the small molecules were obtained by the antechamber module and Hartree-Fock (HF) SCF/6-31G* calculations of the Gaussian 09 software [29, 30]. The small molecule and protein force fields were used for GAFF2 small molecule force field and ff14sb protein force field, respectively [31, 32]. The leap module was used for each system to add hydrogen atoms to the system, a truncated octahedral TIP3P solvent cartridge was added at a distance of 10 Å from the system [33], and Na+/Cl- was added to balance the system charge. A 200 ps ramp-up of the system was performed to slowly increase the system temperature from 0 to 298.15 K. A 500 ps simulation of the NVT (isothermal isomer) tether was performed with the system maintained at 298.15 K. In the case of NPT (isothermal isobaric), equilibrium simulations were performed for the whole system for 500 ps. Finally, 100 ns of NPT (isothermal isobaric) tethering simulations were performed separately. For the simulations, the non-bond truncation distance is set to 10 Å. The Particle mesh Ewald (PME) method is used to calculate the long-range electrostatic interaction [34], the SHAKE method is used to limit the bond length of hydrogen atoms [35], and the Langevin algorithm is used for temperature control [36], where the collision frequency γ is set to 2 ps-[1]. The system pressure is 1 atm, the integration step is 2 fs, and the trajectories are saved at 10 ps intervals for subsequent analysis. The traces were saved at 10 ps intervals for subsequent analysis.
The binding free energies between proteins and ligands for all systems were calculated by the MM/GBSA method [37,38,39,40]. The long-time molecular dynamics simulations may not be conducive to the accuracy of MM/GBSA calculations [38]. Therefore, the MD trajectory of 90–100 ns was used as the calculation in this study with the following equations.
In Eq. (1), \({{\Delta }\text{E}}_{\text{i}\text{n}\text{t}\text{e}\text{r}\text{n}\text{a}\text{l}}\) denotes internal energy、\({{\Delta }\text{E}}_{\text{V}\text{D}\text{W}}\)denotes van der Waals interaction, and \({{\Delta }\text{E}}_{\text{e}\text{l}\text{e}\text{c}}\) denotes electrostatic interaction. The internal energy includes Ebond, Eangle, and Etorsion;\({{\Delta }\text{G}}_{\text{G}\text{B}}\) and \({{\Delta }\text{G}}_{\text{G}\text{A}}\) are collectively referred to as the solventization free energy. Among them, GGB is the polar solvation free energy and GSA is the non-polar solvation free energy. For \({{\Delta }\text{G}}_{\text{G}\text{B}}\), the GB model (igb = 2) developed by Nguyen [41] is used for the calculation. The nonpolar solvation free energy (ΔGSA) is calculated based on the product of surface tension (γ) and solvent accessible surface area (SA), ΔGSA= 0.0072 × ΔSASA [42]. The entropy change is neglected in this study due to high computational resources and low precision; this study was neglected [37, 38].
Gene Set Enrichment Analysis (GSEA)
The Gene Set Enrichment Analysis [43] was performed by “ggplot2”, “limma”, “ggsci”, “org.Hs.eg.db” and “patchwork”. We selected the top 3 genes of molecular docking results for GSEA single gene analysis, and the disease group of GSE15932 (diabetes) was selected for the signature gene set. Enrichment scores (ES) were calculated based on weighted Kolmogorov-Smirnov class statistics, and their magnitude reflects the correlation between gene set and phenotype. A higher ES of a gene set implies a higher likelihood that the gene set is enriched in a specific phenotype [44].
Result
Data collection of disease and drug components
First, to precisely identify the targets of diabetes and AD. We retrieved DEGs by comparing the differential gene expression levels between control and disease groups. GSE15653 selected 5 control and 9 diabetes group samples and analyzed 995 up-regulated and 982 down-regulated genes (Fig. 2a); GSE132903 selected 98 control and 97 AD group samples and analyzed 336 down-regulated genes and 286 up-regulated genes (Fig. 2c). The DEGs of the top 30 up and down-regulated genes (Fig. 2b, d) were also shown with heat maps. 1051 diabetes targets and 1354 AD targets were identified in genecards; 225 diabetes targets and 546 AD targets were identified in OMIM as complementary targets for diabetes and AD.

Screening of diabetes and AD targets. Differential volcano plot showing the gene distribution of disease samples, GSE15653 (a) and GSE132903(b), The heat map shows the top 30 up- and down-regulated DEGs, GSE15653 (c) and GSE132903 (d)
To identify the potentially active chemical components in Chinese medicine. The chemical composition of each herbal drug collected from TCMSP was initially screened using the OB and DL properties of the drugs. The literature was also reviewed and a total of 108 potential components were identified. Among them, SZR and FL both had 4 components, ZM had 11 components, CX had 6 components and GC had 85 components, MOL000359 was common to GC and CX, and MOL000422 was common to ZM and GC. After the uniport transformation of the targets, a total of 253 targets corresponding to the compounds were found in TCMSP. The intersection of 97 targets among drug, diabetes, and AD were obtained by making a Venn diagram (Fig. 3a).

Network pharmacology. a Venn diagram of 97 intersecting targets between drug, diabetes, and AD, (b) D-C-T-D network diagram, gancao (blue green), zhimu (blue-violet), chuangxiong (red), suanzaoren (green), fuling (yellow), and targets (light yellow). c PPI network of 97 intersecting targets. d The top 9 gene is selected MCC from the PPI network
Construction of D-C-T-D networks and PPI networks
These intersection targets and corresponding components were imported into Cytoscape to draw a D-C-T-D network graph, including 212 nodes and 1114 edges (Fig. 3b). Network analysis allows the identification of potentially active compounds for drug therapeutic action. The network was analyzed by Cytoscape, and MOL000098 (degree: 69), MOL000422 (degree: 26), MOL004328 (degree: 19), MOL000392 (degree: 16), MOL000354 (degree: 16), and MOL000497 (degree: 15) were the key compounds (Table 1). The 97 intersecting targets were also imported into the string database to obtain the PPI network graph, with 97 points and 641 edges (Fig. 3c). These results suggest that SZRD may act on Alzheimer’s with diabetes through these compounds, and the existence of a strong association between these targets suggests that SZRD can have a therapeutic effect on Alzheimer’s with diabetes through the ability of multiple compounds and multiple targets thus.
Hub gene extraction and MCODE analysis
Most of the interconnected nodes in a PPI network are considered to be hub genes in the PPI network. According to the PPI network analysis of the Cytohubba plugin in Cytoscape, we listed the top 9 (9.28%) DEGs as the most influential genes (Fig. 3d). The hub gene is namely IL6 (Score: 9.75E + 08), TNF (Score: 9.75E + 08), IL1B (Score: 9.74E + 08), CXCL8 (Score: 9.74E + 08), IL10 (Score: 9.74E + 08), CCL2 (Score: 9.74E + 08), ICAM1 (Score: 9.74E + 08), STAT3 (Score: 9.66E + 08) and IL4 (Score: 9.62E + 08). These hub genes may be potential biomarkers, which may also be new disease treatment strategies. To better understand these targets, we performed MCODE module analysis with the help of Metascape to gain more insight into the degree of target clustering (Fig. 4). The results of MCODE clustering analysis showed that these targets were mainly clustered in pathway in cancer, inflammatory response, AGE-RAGE signaling pathway in diabetic complications, inflammatory response, and response to peptide.

Results of MCODE analysis. a Network diagram of PPI after MCODE clustering analysis. b Network diagram of MCODE clustering separately. c The entries, categories, and corresponding log p-values of each cluster were obtained from the MCODE cluster analysis
GO and KEGG enrichment analysis
R was used to perform GO and KEGG enrichment analysis to identify intersecting genes that share the biological significance and enrichment pathways highlighted in this study. Gene ontology considers gene functions and their components, providing a broad resource of computable knowledge. Gene ontology analysis was performed for three categories (biological processes, cellular components, and molecular functions) and the GO database was selected as the annotation source. KEGG analysis reveals the response of an organism to its intrinsic modifications. It is a modeling technique to demonstrate the interactions between various diseases through underlying molecular or biological processes. Table 2 summarizes the top 10 terms in the categories of biological processes, molecular functions, and cellular components; Table 3 summarizes the top 20 terms in the KEGG pathway. We plotted network diagrams (Fig. 5a, b, c, e) and bubble diagrams (Fig. 5d, f) for the top ten GO entries and KEGG pathways and corresponding targets. These results can reveal that SZRD can function in multiple biological processes and signaling pathways, probably mainly enriched in biological processes such as response to nutrient levels, response to oxidative stress, and cellular response to chemical stress. KEGG enrichment analysis showed that the KEGG enrichment was mainly in multiple pathways such as Fluid shear stress and atherosclerosis, AGE-RAGE signaling pathway in diabetic complications, and Lipid and atherosclerosis.

GO and KEGG enrichment analysis results. network plots between the top 10 pathways for each category in GO enrichment analysis BP (a), CC (b), and MF (c). d Bubble plots of GO enrichment analysis. e Network diagram of the top 10 pathways analyzed by KEGG enrichment. f Bubble plots of KEGG enrichment analysis
We found that through AGE-RAGE signaling pathway in diabetic complications and Fluid shear stress and atherosclerosis, signaling pathways were highly expressed in KEGG enrichment analysis and MCODE analysis, using the “pathview” package for the visualization of the pathways (Fig. 6). From the pathway map, we can find that PI3K/AKT signaling pathway and MAPK signaling pathway form the AGE-RAGE signaling pathway in diabetic complications and Fluid shear stress and atherosclerosis, so we think PI3K/AKT signaling pathway and MAPK signaling pathway may have a strong correlation in these pathways. But the exact mechanism may need further validation.

Distribution of intersecting genes in related pathways. a AGE-RAGE signaling pathway in diabetic complications, (b) Fluid shear stress and atherosclerosis signaling pathway. Red rectangles represent key targets
Molecular docking
Molecular docking is a bioinformatics tool that is the process of finding the best combination of small molecules (ligands) and biomacromolecules (receptors) by intermolecular geometric matching and energy calculation of their patterns and effectiveness. Molecular docking of the first 6 active compounds and 9 hub genes was performed using autodocktools software, and a heat map of the docking results was created using “pheatmeap” (Fig. 7). Then we used PYMOL to visualize the molecular docking results in 3D and discovery studio software to visualize the docking results in 2D and selected the five compounds with the best docking activation performance to the hub gene (Fig. 8). The results showed that CXCL8 had high binding activity to most of the compounds, with licochalcone a, isorhamnetin, kaempferol, quercetin, and formononetin having the strongest docking activity, all with docking activation energies between − 8.5 and − 9.8, which also implies stable binding. From the 2D results, the binding affinity was mainly attributed to hydrogen bonding, Pi-alkyl, pi-cation, and van der Waals forces. We found that these potential compounds have strong binding affinity to the core gene and molecular docking can only predict the binding pattern between the core gene and the potential compound. It should be noted that we still need further experimental validation of these potential compounds and targets is needed.

The molecular docking results of the first 6 potential compounds with 9 hub genes are scored in a heat map (unit: kcal/mol), with red representing high docking activation energy and blue representing low docking activation energy

Visualization of docking results of hub gene with potential compounds, licochalcone a (a), isorhamnetin (b), kaempferol (c), quercetin (d), and formononetin (e) generated with CXCL8 in 3D and 2D, with 3D results on the left and 2D results on the right
Molecular dynamics simulation
Molecular dynamics can be good for discovering the relationship between proteins and ligands. We performed molecular dynamics simulations for CXCL8- licochalcone a, CXCL8- isorhamnetin and CXCL8- kaempferol for 100ns to evaluate the intermolecular motions, trajectories, structures, binding potentials, and conformational changes by analyzing the molecular docking results.
Root-mean-square deviation (RMSD) can reflect the motion process of the complexes, with larger RMSD as well as more intense fluctuations indicating violent motion and, conversely, smooth motion. In conclusion, our study shows that the system maintains a stable RMSD fluctuation under the binding of three small molecules, which means that the small molecules are stable (Fig. 9a). Root Mean Square Fluctuation (RMSF) can respond to the flexibility of the protein during molecular dynamics simulations. about the same, indicating that Isorhamnetin, Kaempferol, and Licochalcone_A binding is stable and does not affect the flexibility of the protein (Fig. 9b). The hydrogen bond is one of the strongest non-covalent binding interactions, and the higher number indicates better binding. how the three groups of CXCL8/Isorhamnetin, CXCL8/Kaempferol, and CXCL8/Licochalcone_A generally had between 1 and 2 hydrogen bonds during the simulation (Fig. 9c). It indicates that hydrogen bonding plays a role in the binding of small molecules and proteins during kinetic simulation.

The molecular dynamics (MD) simulation of the CXCL8- licochalcone a complex, the CXCL8- isorhamnetin complex, and the CXCL8- kaempferol complex. a The RMSD plot of the complexes. b The RMSF plot of the complexes. c The number of hydrogen bonds in the complexes
Based on the trajectories of molecular dynamics simulations, we calculated the binding energy using the MM-GBSA method, which can more accurately reflect the binding effect of small molecules and target proteins. the binding energies of CXCL8/Isorhamnetin, CXCL8/Kaempferol, and CXCL8/Licochalcone_A were − 15.83 ± Negative values indicate that the molecule has a binding affinity to the target protein, and lower values indicate stronger binding. Our calculations indicate that CXCL8/Isorhamnetin, CXCL8/Kaempferol, and CXCL8/Licochalcone_A have binding potential. By energy decomposition, we can see that the main contribution to the binding of licochalcone a, isorhamnetin, and kaempferol to CXCL8 can be van der Waals energy, followed by electrostatic energy and non-polar solvation free energy (Table 4). In conclusion, these results demonstrate the reliability of our molecular docking results and the binding stability of CXCL8 with licochalcone a, isorhamnetin, and kaempferol.
Gene set enrichment analysis
GSEA enrichment analysis can analyze the regulation of genes in the dataset. Molecular docking identified genes that bind strongly to core compounds, such as CXCL8, IL1B, and CCL2. we then subjected these genes to GSEA analysis in GSE15932 to find their effects in the gene set. the GSEA results showed that CXCL8, IL1B and CCL2 affect Graft-versus-host disease, Legionellosis, Primary immunodeficiency, Primary bile acid biosynthesis, Protein export and Non-homologous end-joining signaling pathways (Fig. 10). These results suggest a regulatory role of these genes in diabetes, mainly related to immunity and inflammation.

Enrichment of hub gene in GSE15932 by GSEA. a and b CXCL8, up (a), down (b). c and(d)CCL2, up (c), down (d). e and f IL1B, up (e), down (f)
Discussion
Diabetes not only increases the risk of cerebrovascular disease and stroke but also exacerbates neurodegenerative diseases, particularly AD [45]. Traditional Chinese medicine has gained international recognition over the course of thousands of years, with numerous prescriptions being utilized to treat a wide range of clinical conditions. In Traditional Chinese Medicine, SZRD is a classical prescription known for its sedative and tranquilizing properties, primarily used in the treatment of conditions such as “liver and blood deficiency, heat deficiency, and internal disorders" [46]. Diabetes is categorized as “depression” and “thirst” in Traditional Chinese Medicine and is believed to be primarily related to yin and qi deficiency in the patient’s body, as well as emotional and willpower disorders. Therefore, clinical treatment should focus on relieving liver depression, nourishing yin and generating fluid and tonifying qi and blood. Recent studies have shown that SZRD improves cognitive function in APP/PS1 mice and reduces levels of IL-6, IL-1β, and TNF-α [9]. These findings indicate that SZRD may have therapeutic effects on Alzheimer’s with diabetes, although its mechanism of action remains unclear. Hence, we employed a combined approach of bioinformatics, network pharmacology, and molecular docking to identify the active compounds and potential targets of SZRD in Alzheimer’s with diabetes.
We employed the principles of oral bioavailability (OB) and drug-likeness (DL) for active compound screening, and integrated them with disease targets to identify 97 intersecting targets using the D-C-T-D approach. Among these, quercetin, kaempferol, licochalcone a, isorhamnetin, formononetin, and naringenin were identified as the main core compounds, which have also been demonstrated to exhibit therapeutic effects in both diabetes and AD. Licochalcone a, a naturally occurring specific inhibitor of JNK (c-Jun N-terminal kinase), has been demonstrated to exhibit therapeutic effects in diabetic nephropathy [47]. Isorhamnetin, a regulator of the insulin signaling pathway, has been shown to improve diabetes by mitigating insulin resistance [48]. Formononetin, a naturally occurring isoflavone, acts as a non-classical agonist of PPARγ (peroxisome proliferator-activated receptor gamma), which not only treats Alzheimer’s with diabetes but also enhances fat thermogenesis to reduce obesity [49]. Naringenin has also been demonstrated to possess potent neuroprotective and antidiabetic effects [50]. Furthermore, these compounds highlight the therapeutic potential of SZRD in addressing cognitive impairment associated with diabetes.
We retrieved 1977 DEGs for diabetes and 622 DEGs for AD from the GEO database. After intersecting the targets of complementary targets and drugs, a total of 97 targets were identified. Further, we performed MCC analysis on the PPI network to identify Hub targets including IL6, TNF, IL1B, CXCL8, IL10, CCL2, ICAM1, STAT3, and IL4. Low-grade inflammation, which plays a role in the development and progression of multiple diseases, has been associated with IL6, IL-1B, CXCL8, IL4, TNF, and IL10 in various diseases [51, 52]. CCL2 acts as a ligand for the C-C chemokine receptor CCR2, which is stimulated by inflammation [53]. Meta-analysis demonstrated that ICAM1, a cell adhesion molecule, is elevated in the circulatory system of diabetic patients, and its levels are dose-dependently associated with the risk of diabetes [54]. STAT3 has been proposed as a potential link between inflammation and chronic disease [55]. We observed a strong correlation between these hub genes and inflammation, which is a critical pathological feature of Alzheimer’s with diabetes. These findings imply that SZRD may primarily exert an anti-inflammatory role in treating Alzheimer’s with diabetes at the genetic level.
GO enrichment analysis revealed that SZRD regulates Alzheimer’s with diabetes through biological processes such as response to nutrient levels, oxidative stress, and cellular response to chemical stress. Oxidative stress and inflammation play a role in the development of Alzheimer’s with diabetes. Our KEGG enrichment analysis indicated that these targets were mainly enriched in pathways such as Fluid shear stress and atherosclerosis, AGE-RAGE signaling pathway in diabetic complications, Lipid and atherosclerosis, IL-17 signaling pathway, and TNF signaling pathway. Previous studies have demonstrated that Fluid shear stress and atherosclerosis, as well as the AGE-RAGE signaling pathway in diabetic complications, play a regulatory role in diabetes [56] and are also closely related to inflammation and oxidation [57]. The KEGG network and target analysis revealed the presence of inflammatory factors and oxidative stress-related proteins involved in network regulation, along with the identification of PI3K/AKT and MAPK signaling pathways in the pathway map that could potentially modulate these factors and proteins. The PI3K/Akt signaling pathway is implicated in the modulation of cytokines and plays a crucial role in regulating insulin and cognitive function in AD through the reduction of phosphorylation levels of PI3K, AKT, and mTOR [58]. Previous studies have demonstrated the involvement of PI3K in the regulation of diabetic complications. Furthermore, PI3K/AKT has been shown to play a significant role in diabetic cognitive impairment [59]. mitogen-activated protein kinase (MAPK) is a serine/threonine protein kinase that regulates multiple inflammatory responses [60], including NLRP3 and NF-κB pathways. Studies have shown that RAGE can activate the P38 MAPK/NF-κB signaling pathway, which is implicated in diabetic cognitive dysfunction [61]. Therefore, PI3K/AKT and MAPK may serve as crucial regulators in the pathways of fluid shear stress, atherosclerosis, and the AGE-RAGE signaling pathway in diabetic complications, potentially contributing to SZRD-mediated Alzheimer’s with diabetes.
Molecular docking was employed to evaluate the activity of active compounds (quercetin, kaempferol, licochalcone a, isorhamnetin, formononetin, and naringenin) with hub genes (IL6, TNF, IL1B, CXCL8, IL10, CCL2, ICAM1, STAT3, and IL4). The binding affinities were predominantly <-4.25 kcal/mol, indicating robust interactions between most compounds and hub genes, with kaempferol, licochalcone a, and isorhamnetin exhibiting the strongest docking activities with hub gene CXCL8. Subsequently, we conducted molecular dynamics simulations to further investigate this, and the results further confirmed the strong binding stability of kaempferol, licochalcone a, and isorhamnetin with hub gene CXCL8. Lastly, we performed GSEA analysis on CXCL8, IL1B, and CCL2, three hub genes with significant docking activity, using the results of molecular docking. The GSEA analysis further demonstrated the pivotal roles of CXCL8, IL1B, and CCL2 in diabetes through immune and inflammatory mechanisms. In conclusion, our study elucidates the potential role of active compounds and hub genes of SZRD in the treatment of Alzheimer’s with diabetes.
Limitation
However, there are some limitations of the study in this paper. First, from the perspective of Traditional Chinese Medicine, suanzaoren is considered the core component of this formula. Most of the predicted compounds identified in our study belong to gancao, rather than suanzaoren, which raises concerns about the reliability of the prediction. Secondly, the active compounds of SZRD remain unidentified and could be compensated by additional research methods such as LC/MS, metabolomics, and pharmacokinetics. Finally, validation through animal and cellular experiments was not conducted due to constraints such as time limitations, which could be considered in future research. Our experiments mainly illustrate the preliminary therapeutic effect of SZRD on Alzheimer’s with diabetes, while our results predict the key role played by these active ingredients.
Conclusion
In summary, this study applied bioinformatics, network pharmacology, molecular docking, and molecular dynamics simulation to predict the pharmacological effects and molecular mechanisms of SZRD in Alzheimer’s with diabetes. Licochalcone A, isorhamnetin, and kaempferol have the potential to serve as the primary active ingredients in SZRD treatment. The results of molecular dynamics simulations also suggest a strong association between the hub gene and Licochalcone A, isorhamnetin, and kaempferol. Our results provide a theoretical basis for subsequent experimental verification.
Availability of data and materials
Data will be made available on request.
Abbreviations
- TCM:
-
Traditional Chinese Medicine
- SZRD:
-
Suanzaoren Decoction
- AD:
-
Alzheimer's disease
- DEG:
-
Differentially expressed genes
- GSEA:
-
Gene Set Enrichment Analysis
- Suanzaoren, SZR:
-
Ziziphus jujuba Mill
- Gancao, GC:
-
Glycyrrhiza uralensis Fisch
- Zhimu, ZM:
-
Anemarrhena asphodeloides Bunge
- Fuling, FL:
-
Poria cocos (Schw.) Wolf
- Chuanxiong, CX:
-
Ligusticum acuminatum Franch
- GEO:
-
Gene Expression Omnibus
- BC:
-
Betweenness centrality
- CC:
-
Closeness centrality
- ASPL:
-
Average shortest path length
- MCC:
-
Maximum population centrality
- D-C-T-D:
-
Drug-compound-target-disease
- PPI:
-
Protein-protein interaction
- BP:
-
Biological process
- CC:
-
Cellular component
- MF:
-
Molecular function
- HF:
-
Hartree-Fock
- OB:
-
Oral bioavailability
- DL:
-
Drug similarity
- PME:
-
Particle mesh Ewald
- ES:
-
Enrichment scores
References
Gregory GA, Robinson TIG, Linklater SE, Wang F, Colagiuri S, de Beaufort C, Donaghue KC, Magliano DJ, Maniam J, Orchard TJ, et al. Global incidence, prevalence, and mortality of type 1 diabetes in 2021 with projection to 2040: a modelling study. Lancet Diabetes Endocrinol. 2022;10(10):741–60. https://doi.org/10.1016/S2213-8587(22)00218-2PubMed.
Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB, Stein C, Basit A, Chan JCN, Mbanya JC, et al. IDF diabetes atlas: global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract. 2022;183: 109119. https://doi.org/10.1016/j.diabres.2021.109119PubMed.
Hurd MD, Martorell P, Delavande A, Mullen KJ, Langa KM. Monetary costs of dementia in the United States. N Engl J Med. 2013;368(14):1326–34. https://doi.org/10.1056/NEJMsa1204629PubMed.
Xue M, Xu W, Ou Y-N, Cao X-P, Tan M-S, Tan L, Yu J-T. Diabetes mellitus and risks of cognitive impairment and dementia: a systematic review and meta-analysis of 144 prospective studies. Ageing Res Rev. 2019;55: 100944. https://doi.org/10.1016/j.arr.2019.100944PubMed.
Biessels GJ, Despa F. Cognitive decline and dementia in diabetes mellitus: mechanisms and clinical implications. Nat Rev Endocrinol. 2018;14(10):591–604. https://doi.org/10.1038/s41574-018-0048-7PubMed.
Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P. Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol. 2006;5(1):64–74 (PubMed).
Koekkoek PS, Kappelle LJ, van den Berg E, Rutten GEHM, Biessels GJ. Cognitive function in patients with Diabetes Mellitus: guidance for daily care. Lancet Neurol. 2015;14(3):329–40. https://doi.org/10.1016/S1474-4422(14)70249-2PubMed.
Liu J-X, Li J-H, Du C-H, Yan Y. [Metabonomic study of biochemical changes in serum of PCPA-induced insomnia rats after treatment with Suanzaoren Decoction]. Zhongguo Zhong Yao Za Zhi. 2022;47(6):1632–41. https://doi.org/10.19540/j.cnki.cjcmm.20211204.701PubMed.
Long Q-H, Wu Y-G, He L-L, Ding L, Tan A-H, Shi H-Y, Wang P. Suan-Zao-Ren decoction ameliorates synaptic plasticity through inhibition of the Aβ deposition and JAK2/STAT3 signaling pathway in AD model of APP/PS1 transgenic mice. Chin Med. 2021;16(1):14. https://doi.org/10.1186/s13020-021-00425-2PubMed.
Zhou YP, Zhang JQ. Oral baicalin and liquid extract of licorice reduce sorbitol levels in red blood cell of diabetic rats. Chin Med J (Engl). 1989;102(3):203–6 (PubMed).
Chen J, Wan L, Zheng Q, Lan M, Zhang X, Li Y, Li B, Li L. Structural characterization and in vitro hypoglycaemic activity of glucomannan from anemarrhena asphodeloides bunge. Food Funct. 2022;13(4):1797–807. https://doi.org/10.1039/d1fo03010hPubMed.
Hopkins AL. Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol. 2008;4(11):682–90. https://doi.org/10.1038/nchembio.118PubMed.
Wang X, Wang Z-Y, Zheng J-H, Li S. TCM network pharmacology: a new trend towards combining computational, experimental and clinical approaches. Chin J Nat Med. 2021;19(1). https://doi.org/10.1016/S1875-5364(21)60001-8.
Du L, Geng C, Zeng Q, Huang T, Tang J, Chu Y, Zhao K. Dockey: a modern integrated tool for large-scale molecular docking and virtual screening. Brief Bioinform. 2023;24(2): bbad047. https://doi.org/10.1093/bib/bbad047.
Hildebrand PW, Rose AS, Tiemann JKS. Bringing molecular dynamics simulation data into view. Trends Biochem Sci. 2019;44(11):902–13. https://doi.org/10.1016/j.tibs.2019.06.004PubMed.
Ru J, Li P, Wang J, Zhou W, Li B, Huang C, Li P, Guo Z, Tao W, Yang Y, et al. TCMSP: a database of systems pharmacology for drug discovery from herbal medicines. J Cheminform. 2014;6: 13. https://doi.org/10.1186/1758-2946-6-13PubMed.
Wang Y, Yu W, Shi C, Jiao W, Li J, Ge J, Hong Y, Shi G. Network Pharmacology of Yougui Pill Combined with Buzhong Yiqi Decoction for the treatment of sexual dysfunction. Evid Based Complement Alternat Med. 2019;2019–1243743. https://doi.org/10.1155/2019/1243743. PubMed.
Hou F, Yu Z, Cheng Y, Liu Y, Liang S, Zhang F. Deciphering the pharmacological mechanisms of Scutellaria baicalensis Georgi on oral leukoplakia by combining network pharmacology, molecular docking and experimental evaluations. Phytomedicine. 2022;103: 154195. https://doi.org/10.1016/j.phymed.2022.154195PubMed.
Xiong G, Wu Z, Yi J, Fu L, Yang Z, Hsieh C, et al. ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021;49(W1). https://doi.org/10.1093/nar/gkab255.
Shen J, Zhu B. Integrated analysis of the gene expression profile and DNA methylation profile of obese patients with type 2 Diabetes. Mol Med Rep. 2018;17(6):7636–44. https://doi.org/10.3892/mmr.2018.8804PubMed.
Gan X-X, Zhong L-K, Shen F, Feng J-H, Li Y-Y, Li S-J, Cai W-S, Xu B. Network Pharmacology to explore the Molecular mechanisms of for treating Hashimoto’s Thyroiditis. Front Pharmacol. 2021;12: 700896. https://doi.org/10.3389/fphar.2021.700896PubMed.
Liu J, Liu J, Tong X, Peng W, Wei S, Sun T, Wang Y, Zhang B, Li W. Network pharmacology prediction and molecular docking-based strategy to discover the potential pharmacological mechanism of huai hua san against ulcerative colitis. Drug Des Devel Ther. 2021;15:3255–76. https://doi.org/10.2147/DDDT.S319786PubMed.
Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, Doncheva NT, Legeay M, Fang T, Bork P, et al. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021;49(D1):D605-612. https://doi.org/10.1093/nar/gkaa1074PubMed.
Chin C-H, Chen S-H, Wu H-H, Ho C-W, Ko M-T, Lin C-Y. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;8(Suppl 4): 11. https://doi.org/10.1186/1752-0509-8-S4-S11PubMed.
Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, Chanda SK. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10(1):1523. https://doi.org/10.1038/s41467-019-09234-6PubMed.
Yang X, Liu Y, Gan J, Xiao Z-X, Cao Y. FitDock: protein-ligand docking by template fitting. Brief Bioinform. 2022;23(3): bbac087. https://doi.org/10.1093/bib/bbac087.
Shan C, Ji X, Wu Z, Zhao J. Network pharmacology combined with GEO database identifying the mechanisms and molecular targets of Polygoni Cuspidati Rhizoma on peri-implants. Sci Rep. 2022;12(1):8227. https://doi.org/10.1038/s41598-022-12366-3PubMed.
Salomon-Ferrer R, Case DA, Walker RC. An overview of the Amber biomolecular simulation package. Wiley Interdiscip Rev Comput Mol Sci. 2013;3(2):198–210. https://doi.org/10.1002/wcms.1121.
Frisch M, Trucks G, Schlegel H, Scuseria G, Robb M, Cheeseman J, et al. Gaussian 09 (Revision D.01). 2009.
Wang J, Wang W, Kollman PA, Case DA. Antechamber: an accessory software package for molecular mechanical calculations. J Am Chem Soc. 2001;222:U403.
Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25(9):1157–74.
Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. ff14SB: improving the Accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput. 2015;11(8):3696–713. https://doi.org/10.1021/acs.jctc.5b00255.
Mark P, Nilsson L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J Phys Chem A. 2001;105(43):9954–60. https://doi.org/10.1021/jp003020w.
Sagui C, Darden TA. Molecular dynamics simulations of biomolecules: long-range electrostatic effects. Annu Rev Biophys Biomol Struct. 1999;28(1):155–79. https://doi.org/10.1146/annurev.biophys.28.1.155.
Kräutler V, Van Gunsteren WF, Hünenberger PH. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J Comput Chem. 2001;22(5):501–8. https://doi.org/10.1002/1096-987X(20010415)22:5%3c501::AID-JCC1021%3e3.0.CO;2-V.
Larini L, Mannella R, Leporini D. Langevin stabilization of molecular-dynamics simulations of polymers by means of quasisymplectic algorithms. J Chem Phys. 2007;126(10):104101. https://doi.org/10.1063/1.2464095. (From NLM).
Chen Y, Zheng Y, Fong P, Mao S, Wang Q. The application of the MM/GBSA method in the binding pose prediction of FGFR inhibitors. Phys Chem Chem Phys. 2020;22(17):9656–63. https://doi.org/10.1039/d0cp00831a.
Hou T, Wang J, Li Y, Wang W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J Chem Inf Model. 2011;51(1):69–82. https://doi.org/10.1021/ci100275a. (From NLM).
Genheden S, Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov. 2015;10(5):449–61. https://doi.org/10.1517/17460441.2015.1032936.
Rastelli G, Rio AD, Degliesposti G, Sgobba M. Fast and accurate predictions of binding free energies using MM-PBSA and MM‐GBSA. J Comput Chem. 2010;31(4):797–810.
Nguyen H, Roe DR, Simmerling C. Improved generalized born Solvent Model parameters for protein simulations. J Chem Theory Comput. 2013;9(4):2020–34. https://doi.org/10.1021/ct3010485.
Weiser J, Shenkin PS, Still WC. Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO). J Comput Chem. 1999;20(2):217–30.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50 (PubMed).
Yang L, Peng X, Li Y, Zhang X, Ma Y, Wu C, Fan Q, Wei S, Li H, Liu J. Long non-coding RNA HOTAIR promotes exosome secretion by regulating RAB35 and SNAP23 in hepatocellular carcinoma. Mol Cancer. 2019;18(1):78. https://doi.org/10.1186/s12943-019-0990-6PubMed.
Arnold SE, Arvanitakis Z, Macauley-Rambach SL, Koenig AM, Wang H-Y, Ahima RS, Craft S, Gandy S, Buettner C, Stoeckel LE, et al. Brain insulin resistance in type 2 Diabetes and Alzheimer Disease: concepts and conundrums. Nat Rev Neurol. 2018;14(3):168–81. https://doi.org/10.1038/nrneurol.2017.185PubMed.
Liu Y-Y, Yu L-H, Zhang J, Xie D-J, Zhang X-X, Yu J-M. Network pharmacology-based and molecular docking-based analysis of suanzaoren decoction for the treatment of parkinson’s disease with sleep disorder. Biomed Res Int. 2021;2021: 1752570. https://doi.org/10.1155/2021/1752570.
Luo Z, Li T, Gao Q, Chen Y, Su G, Zhao Y. Impact of licochalcone A on the progression of diabetic Nephropathy in type 2 diabetes mellitus of C57BL/6 mice. Food Funct. 2021;12(21):10676–89. https://doi.org/10.1039/d1fo01630jPubMed.
Matboli M, Saad M, Hasanin AH, Saleh A, Baher L, Bekhet W, Eissa MM. New insight into the role of isorhamnetin as a regulator of insulin signaling pathway in type 2 Diabetes Mellitus rat model: Molecular and computational approach. Biomed Pharmacother. 2021;135: 111176. https://doi.org/10.1016/j.biopha.2020.111176PubMed.
Nie T, Zhao S, Mao L, Yang Y, Sun W, Lin X, Liu S, Li K, Sun Y, Li P, et al. The natural compound, formononetin, extracted from Astragalus Membranaceus increases adipocyte thermogenesis by modulating PPARγ activity. Br J Pharmacol. 2018;175(9):1439–50. https://doi.org/10.1111/bph.14139PubMed.
Singh S, Sharma A, Monga V, Bhatia R. Compendium of naringenin: potential sources, analytical aspects, chemistry, nutraceutical potentials and pharmacological profile. Crit Rev Food Sci Nutr. 2022. https://doi.org/10.1080/10408398.2022.2056726PubMed.
Lehrskov LL, Christensen RH. The role of interleukin-6 in glucose homeostasis and lipid metabolism. Semin Immunopathol. 2019;41(4):491–9. https://doi.org/10.1007/s00281-019-00747-2PubMed.
Lu J, Liu J, Li L, Lan Y, Liang Y. Cytokines in type 1 Diabetes: mechanisms of action and immunotherapeutic targets. Clin Transl Immunology. 2020;9(3): e1122. https://doi.org/10.1002/cti2.1122PubMed.
Lin M, Yiu WH, Wu HJ, Chan LYY, Leung JCK, Au WS, Chan KW, Lai KN, Tang SCW. Toll-like receptor 4 promotes tubular inflammation in diabetic Nephropathy. J Am Soc Nephrol. 2012;23(1):86. https://doi.org/10.1681/ASN.2010111210.
Qiu S, Cai X, Liu J, Yang B, Zügel M, Steinacker JM, Sun Z, Schumann U. Association between circulating cell adhesion molecules and risk of type 2 Diabetes: a meta-analysis. Atherosclerosis. 2019;287:147–54. https://doi.org/10.1016/j.atherosclerosis.2019.06.908PubMed.
Gupta SC, Kunnumakkara AB, Aggarwal S, Aggarwal BB. Inflammation, a double-edge Sword for Cancer and other Age-Related Diseases. Front Immunol. 2018;9: 2160. https://doi.org/10.3389/fimmu.2018.02160PubMed.
Hung T-C, Zhao N, Huang C, Liu S, Liu T, Huang W, Xu X, Ji Z-L, Yang S. Exploring the mechanism of PingTang No.5 capsule on nonalcoholic fatty liver disease through network pharmacology and experimental validation. Biomed Pharmacother. 2021;138: 111408. https://doi.org/10.1016/j.biopha.2021.111408PubMed.
Cheng H, Zhong W, Wang L, Zhang Q, Ma X, Wang Y, Wang S, He C, Wei Q, Fu C. Effects of shear stress on vascular endothelial functions in Atherosclerosis and potential therapeutic approaches. Biomed Pharmacother. 2023;158: 114198. https://doi.org/10.1016/j.biopha.2022.114198PubMed.
Arvanitakis Z, Wang H-Y, Capuano AW, Khan A, Taïb B, Anokye-Danso F, Schneider JA, Bennett DA, Ahima RS, Arnold SE. Brain insulin signaling, alzheimer disease pathology, and cognitive function. Ann Neurol. 2020;88(3):513–25. https://doi.org/10.1002/ana.25826PubMed.
Wu Y, Wu C, Ye L, Wang B, Yuan Y, Liu Y, Zheng P, Xiong J, Li Y, Jiang T, et al. Exogenous fibroblast growth factor 1 ameliorates diabetes-induced cognitive decline via coordinately regulating PI3K/AKT signaling and PERK signaling. Cell Commun Signal. 2020;18(1):81. https://doi.org/10.1186/s12964-020-00588-9PubMed.
Ronkina N, Gaestel MMAPK-A. Protein kinases: servant or Partner? Annu Rev Biochem. 2022;91:505–40. https://doi.org/10.1146/annurev-biochem-081720-114505PubMed.
Zhou X-Y, Ying C-J, Hu B, Zhang Y-S, Gan T, Zhu Y-D, Wang N, Li A-A, Song Y-J. Receptor for advanced glycation end products aggravates cognitive deficits in type 2 Diabetes through binding of C-terminal AAs 2–5 to mitogen-activated protein kinase kinase 3 (MKK3) and facilitation of MEKK3-MKK3-p38 module assembly. Aging Cell. 2022;21(2):e13543. https://doi.org/10.1111/acel.13543PubMed.
Acknowledgements
This work was supported by grants from the Hubei University of Science and Technology (2022TNB02, 2022TNB10) to FC or RYZ, The Department of Education of Hubei Province (T201921, B2022190) to FC or RYZ, The Natural Science Foundation of Hubei Province (2023AFD111) to FC, The Natural Science Foundation of xianning(2023ZRKX102) and The Xiannning Central Hospital Fund project(2021XYB033) to XMG.
Author information
Authors and Affiliations
Contributions
Tao Chen and Yining Lei: Writing. Manqin Li: Methodology, Xinran Liu and Lu Zhang: Formal analysis, Fei Cai - Project administration, Funding acquisition, Xiaoming Gong and Ruying Zhang: Writing – original draft, Funding acquisition.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, T., Lei, Y., Li, M. et al. Network pharmacology to unveil the mechanism of suanzaoren decoction in the treatment of alzheimer’s with diabetes. Hereditas 161, 2 (2024). https://doi.org/10.1186/s41065-023-00301-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41065-023-00301-z