
Abstract
Background
Breast cancer has remained the most common malignancy in women over the past two decades. As lifestyle and living environments have changed, alterations to the disease spectrum have inevitably occurred in this time. As molecular profiling has become a routine diagnostic and objective indicator of breast cancer etiology, we analyzed changes in gene expression in breast cancer populations over two decades using The Cancer Genome Atlas database.
Methods
We performed Heatmap and Venn diagram analyses to identify constantly up- and down-regulated genes in breast cancer patients of this cohort. We used Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses to visualize associated functional pathways.
Results
We determined that three oncogenes, PD-L2, ETV5, and MTOR and 113 long intergenic non-coding RNAs (lincRNAs) were constantly up-regulated, whereas two oncogenes, BCR and GTF2I, one tumor suppression gene MEN1, and 30 lincRNAs were constantly down-regulated. Up-regulated genes were enriched in “focal adhesion” and “PI3K-Akt signaling” pathways, etc., and down-regulated genes were significantly enriched in “metabolic pathways” and “viral myocarditis”. Eight up-regulated genes exhibited doubled or higher expression and the expression of three down-regulated genes was halved or lowered and correlated with long-term survival.
Conclusions
In this study, we found that gene expression and molecular pathway enrichments are constantly changing with time, importantly, some altered genes were associated with prognostics and are potential therapeutic targets, suggesting that the current molecular subtyping system must be updated to keep pace with this dynamic change.
Similar content being viewed by others

Introduction
Globally, breast cancer has the highest incidence among all cancers, surpassing lung cancer. In 2020, this disease had an estimated 2.3 million new cases, representing approximately 11.7% of all new cancers [1]. Breast cancer incidence rates are increasing annually [2] and may be due to several key carcinogenic and breast cancer progression factors including, hormonal risk factors (early menarche, late menopause, advanced age at first birth, fewer children being born, lower breastfeeding rates, hormone therapy for the menopause, oral contraceptive use, Vitamin D, and thyroid hormone deficiency), lifestyle risk factors (alcohol intake, excess body weight, physical inactivity, smoking, and antibiotic use), genetic factors (family history of disease and high-penetrance genes), environmental factors (elevated reactive oxygen species levels, higher airborne heavy metals, synthetic chemicals, and radiation), and increased screening [2,3,4,5,6,7,8,9].
Breast cancer detection and intervention at early stages is key in improving prognoses and reducing mortality rates. In the past two decades, researchers have used several conventional and novel breast cancer diagnostic approaches, including mammography, magnetic resonance imaging, ultrasound, biopsies, serum screening for (microRNAs) miRNAs, blood-based proteomics, biomarker analyses, and biosensor technologies [10,11,12]. Based on evidence-based medicine, the comprehensive treatment of breast cancer primarily involves surgery combined with chemotherapy, endocrine therapy, radiation therapy, and targeted therapies [11,12,13]. In recent years, thanks to advances in genetic sequencing techniques, management strategies for malignant tumors have entered a new era of molecular medicine and precise treatment [14,15,16]. Molecular classification, targeted therapy, and immunotherapy approaches aimed at specific genes have considerably ameliorated treatment responses, overall survival (OS), and disease-free survival rates in patients with the disease [17,18,19,20]. However large-scale prospective studies comprising thousands of individuals can take 5–10 years to reach definitive conclusions, the lagging-behind findings have some limitations and defects [21,22,23].
Guidelines on breast cancer screening and diagnosis strategies exert profound effects on breast cancer diagnostics and treatment. One particular, pressing issue relates to whether breast cancer patients diagnosed today are identical or similar to those diagnosed decades ago in terms of clinicopathological characteristics and molecular biological features. This concept is not unusual and is seen in other disciplines such as infectious diseases and climate adaptation. As time progresses, the spectrum of diseases threatening human health is constantly, and indeed, inevitably changing. Globally, at the beginning of the twentieth century, infectious and parasitic diseases were the leading cause of death, however, this status has changed to chronic and degenerative diseases [24, 25]. Climate change is also associated with changes in infectious disease epidemiology; it is predicted that populations at risk for diarrheal disease, malnutrition, and malaria will increase if global warming continues [26,27,28]. Similar studies have been performed for breast cancer; the incidence of estrogen receptor (ER) positive breast cancer has increased slightly for nearly 20 years [29,30,31]. Database analyses have shown that the risk from different types of breast cancer has varied in women of different ages and ethnicities, and has changed over time and not remained static. Yet, gene profiles reflecting breast cancer changes over time have not been reported, therefore, are contemporary gene expression profiles for breast cancer consistent with profiles from 10 or 20 years ago? This question has serious implications for drug development, screening, and therapeutic strategies, therefore, scientists and clinicians rethink and redefine the value of long-standing evidence-based guidelines in guiding clinical practice for emerging diseases. To address this knowledge gap, we used The Cancer Genome Atlas (TCGA) database to generate a preliminary analysis.
Results
Up- and down-regulated genes in breast cancer patients
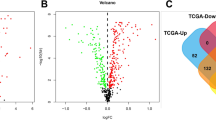
We identified 524 up-regulated and 215 down-regulated genes in 1102 patients. Patients diagnosed with breast cancer between 1988 and 2011 were classified into eight groups according to the year of diagnosis. A heatmap of the top 50 up-regulated and 50 down-regulated genes from eight groups was generated (Fig. 1A; red = up-regulated and blue = down-regulated genes). When we compared the 2011 group with the initial 1998–2000 group, the top five up-regulated genes with the largest log2 fold-change in expression were; AC007728.3, AC097460.1, AC010542.4, USP50, and BX276092.9, at 2.5, 2.3, 2.2, 2.1, and 2.0, respectively. The top five down-regulated genes with the largest log2 fold-change in expression were; C1QTNF9, AC011479.1, MTND4LP30, KRTDAP, and AP000251.1, at 1.8, 1.7, 1.3, 1.2, and 1.1, respectively. We observed two oncogenes BCR and GTF2I, one tumor suppression gene (TSG), MEN1, and 30 long intergenic non-coding RNAs (lincRNAs) in down-regulated genes (Fig. 1B). Notably, the log2 fold-change in BCR expression was 0.2, with a significant Kaplan-Meier P-value of 0.02. We also identified three oncogenes, PD-L2, ETV5, and MTOR, and 113 lincRNAs in up-regulated genes (Fig. 1C). Likewise, the log2 fold-change in PD-L2 expression was 0.9, but with a borderline significant Kaplan-Meier P-value of 0.06. Additionally, we analyzed four genotyping groups. In 232 patients in the Luminal A group, 665 up-regulated and 553 down-regulated genes were identified. CST1 displayed the largest log2 fold-change in up-regulated expression (3.1), and MPPED1 had the second-largest log2 fold-change decrease at 2.9. The up-regulated genes in Luminal B (125 patients), basal-like (101 patients), and HER2-enriched (58 patients) groups were 637, 668, and 500, respectively, and the highest log2 fold-change genes were IGHV3-20 (8.8), NDUFA5P11 (5.8), and HNRNPA1P26 (6.4), respectively. Also, down-regulated genes in Luminal B, basal-like, and HER2-enriched groups were 547, 800, and 615, respectively; the highest log2 fold-change genes were PLA2G3 (4.2), TRAV18 (5.0), and AL390294.1 (9.7), respectively (Table 1).

Identification of altered genes and associated KEGG analyses from eight groups. A Heatmap showing the top 50 up-regulated and top 50 down-regulated genes from the eight groups. Red = up-regulated; Blue = down-regulated. The expression intensity value is derived from gene expression levels using R software analysis. B Venn diagram showing shared genes between down-regulated genes, recognized oncogenes, lincRNAs, and TSGs. C Venn diagram showing shared genes between up-regulated genes, recognized oncogenes, lincRNAs, and TSGs. D KEGG pathway results of the up-regulated genes. The size of each circle represents the gene number in the corresponding pathway, which is proportional to the circles in the caption. E KEGG pathway results for down-regulated genes; the x-axis represents fold enrichment, different colors represent −log10 (P-value), and circle sizes represent gene numbers in a specific pathway. Abbreviations: lincRNA, long intergenic non-coding RNA; TSG, tumor suppressor gene; KEGG, Kyoto Encyclopedia of Genes and Genomes
Significantly enriched Kyoto encyclopedia of genes and genomes (KEGG) pathways
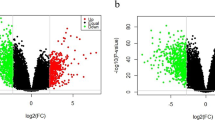
Up-regulated and down-regulated genes were uploaded separately. Up-regulated genes were enriched in 19 pathways, including “focal adhesion”, “PI3K-Akt signaling”, “NOD-like receptor signaling”, “ECM-receptor interaction”, “Toll-like receptor signaling”, etc. Down-regulated genes were significantly enriched in two pathways; “metabolic” and “viral myocarditis” (Fig. 1D, E). The overlapping gene sets in pathways were ITGB1, ITGA4, ACTN1, ROCK1, MTOR, CD80, etc. Interestingly, 10 pathways were enriched in the Luminal A group; “PI3K-Akt signaling” had 20 up-regulated genes, most significantly (p < 0.001, Fig. 2A, B). “ECM-receptor interaction” was immediately followed (p = 0.006). The Luminal B group had seven enriched pathways containing up-regulated genes and 11 pathways containing down-regulated genes (Fig. 2C, D). These encompassed “phagosome”, “platelet activation”, “osteoclast differentiation”, “oxytocin signaling”, “tryptophan metabolism”, “histidine metabolism”, “lysine degradation”, “β-alanine metabolism”, etc. As shown (Fig. 2E, F), 21 enriched pathways were identified in the basal-like group containing up-regulated genes and 15 pathways containing down-regulated genes. For instance, “Ras signaling”, “metabolic”, “insulin signaling”, “thyroid hormone signaling”, “neurotrophy signaling”, “HIF-1 signaling”, “primary immunodeficiency”, and “type I diabetes mellitus”. Furthermore, the HER2-enriched group had five enriched pathways containing up-regulated genes and two pathways containing down-regulated genes (Fig. 2G, H). The “AMPK signaling” and “N-glycan biosynthesis” pathways were the most interesting, with eight up-regulated and four down-regulated genes, respectively.

KEGG pathway analyses of four intrinsic subtypes containing up-regulated and down-regulated genes. A Up-regulated genes in the Luminal A group; B Down-regulated genes in the Luminal A group; C Up-regulated genes in the Luminal B group; D Down-regulated genes in the Luminal B group; E Up-regulated genes in the basal-like group; F Down-regulated genes in the basal-like group; G Up-regulated genes in the HER2-enriched group; H Down-regulated genes in the HER2-enriched group; the x-axis indicates fold enrichment, different colors represent −log10 (P-value), and circle sizes represent gene numbers in a specific pathway. Abbreviations: KEGG, Kyoto Encyclopedia of Genes and Genomes
Survival analysis and expression trends of hub genes
As shown in Fig. 3, eight up-regulated and three down-regulated genes were recognized as hub genes, which satisfied the following conditions; they did not belong to lincRNAs, they had a Kaplan-Meier P-value < 0.05, they had a log2 fold-change in expression > 1 when comparing the 2011 group with the 1998–2000) group, one drop allowed but the change in log2 expression less than one-third of the total change (2011 vs. 1988-2000). As shown in Fig. 4, elevated WFIKKN2, SNORA55, C1QTNF9, and DUSP26 expression displayed significantly improved OS rates and longer median survival times. Also, lower HSP90AA4P, HADHAP1, HADHAP2, and RN7SL738P expression significantly extended patients’ lifespan. Moreover, we hypothesized that USP50, IGLC6, and NACA2 genes, which had log2 fold-expression increases of 2.1, 1.8, and 1.0, respectively, showed potential to become novel clinical outcome predictors and therapeutic targets. Also, the number of people with higher expression had been increasing by about 10% in the last 20 years. In the same way, C1QTNF9 and DUSP26 had log2 fold-decreases in the expression of 1.8 and 1.1 (Fig. 3C), and the number of people with higher expression had been decreasing by about 10% in the last 20 years (Fig. 3D), respectively.

Line charts of log2 fold-changes in the expression of hub up-regulated (A) and down-regulated genes (C) in the eight groups in the TCGA-BRCA database. Line charts of the proportion of high expression population of hub up-regulated (B) and down-regulated genes (D). Abbreviations: TCGA = The Cancer Genome Atlas; BRCA = breast cancer

Kaplan-Meier plots of high and low groups stratified by expression values of hub up-regulated (A) and down-regulated genes (B); blue lines = high expression groups and red lines = low expression groups
Discussion
Over the past 20 years, we observed that > 700 genes had changed and were enriched in “PI3K-Akt signaling”, “ECM-receptor interaction” and “Toll-like receptor signaling”, etc. In different molecular disease groups, enriched pathways containing up-and down-regulated genes were different. For example, “PI3K-Akt signaling” in Luminal A, “phagosome” in Luminal B, “Ras signaling” in basal-like, and “AMPK signaling” in the HER2-enriched group. In addition, 11 genes were > 2-fold altered, were associated with a degree of survival prognosis (p < 0.05), and potentially functioned as therapeutic targets.
Precision medicine has become an essential part of cancer treatment. Targeted molecular therapies and immunotherapies are rapidly moving toward an era of bespoke, precision medicine. Endocrine therapy for ER-positive patients in the 1980s [32, 33] and trastuzumab treatment for HER2-positive patients at the start of this century [34, 35] inaugurated targeted therapies for solid tumors. Surgery, radiotherapy, endocrine therapy, chemotherapy, and/or targeted therapies based on molecular subtyping have also paved the way for “precision medicine” for breast cancer. Additionally, risk prediction models, including the 21-gene assay (Oncotype DX Recurrence Score) and 70-gene assay (commercially known as Mammaprint) have become familiar in clinical settings to provide guidelines for systemic chemotherapy efficacy, and also endocrine therapy which may de-escalate chemotherapy [36,37,38]. Similarly, the inception of gene profiling and next-generation sequencing has meant precision medicine is now closer to clinical practice. Specifically, for ER-positive patients with endocrine therapy resistance, omics-data studies have uncovered mechanisms underpinning “CDK4/6 signaling” and “PI3K-Akt signaling” implicated in tumorigenesis and drug resistance. Similarly, prospective clinical trials also confirmed that the CDK4/6 inhibitor, palbociclib, the mTOR inhibitor, everolimus, and the PI3K inhibitor, buparlisib may improve progression-free survival (PFS) in patients with advanced breast cancer [39,40,41,42]. Also, “MAPK signaling” and “PI3K-Akt signaling” activation are closely associated with tumor cell proliferation in HER2-positive patients. Several clinical trials have explored the efficacy of PI3K inhibitors and tyrosine kinase inhibitors in overcoming resistance to anti-HER2 therapy [43, 44]. In triple-negative breast cancer, immune checkpoint inhibitors may have clinical applications due to “Ras signaling” activation and the elevated expression of immune-related genes such as PD-1, PD-L1, and CTLA-4 [45, 46]. Also, the poly ADP-ribose polymerase (PARP) inhibitors, Olaparib, and talazaparib can prolong PFS and improve patient quality of life in metastatic breast cancer caused by germline BRCA mutations [47,48,49].
We observed that USP50, GPR174, HADHAP2, NACA2, and IGFBPL1 showed large expression changes, significant Kaplan-Meier P values, and increasing proportions in the population. We propose these molecules may serve as potential breast cancer therapeutic targets in the future. Aressy et al. proposed that USP50 repressed activation of DNA damage checkpoints via an HSP90-dependent mechanism, leading to tumors [50]. Smith et al. reported that IGFBP-rP1 and IGFBPL1 expression was regulated by aberrant hypermethylation in breast cancer pathogenesis and that these genes may be beneficial in clinical practice [51]. In the Luminal A group, CST1 exhibited the largest log2 fold expression increase (3.1); a previous study suggested CST1 may function as a significant prognostic indicator and breast cancer therapeutic target [52]. Also, ERBB4 expression exhibited a log2 fold-decrease of 3.7 in the HER2-enriched group, therefore ERBB4 overexpression could have biological and prognostic significance for breast cancer [53].
Interestingly, 113 lincRNAs (21.6%) were up-regulated and 30 (14.0%) down-regulated in our study. Previous research indicated that lincRNAs regulate gene expression at epigenetic and transcription levels, and when the expression is altered, they promote cancer initiation and metastasis. Currently, several lincRNAs are significantly correlated with a cancer diagnosis, prognosis, and the therapeutic development of multitype cancers [54,55,56]. Our data indicated that several lincRNAs could function as potential prognostic biomarkers and have important clinical value, e.g., RFPL1S, ADAMTS9-AS2, IBA57-AS1, and MYOSLID are up-regulated lincRNAs [57,58,59,60,61,62] and MORF4L2-AS1, LINC01278, and LINC00562 [63,64,65,66,67] are down-regulated. Importantly, all are related to the occurrence and development of several tumors by modulating “PI3K-Akt signaling”, “interferon type II signaling” and the expression of particular genes.
We also identified considerable changes in the “Staphylococcus aureus”, “Salmonella” and, “pathogenic Escherichia coli” infection pathways, which we suspect may be related to antibiotics overuse. Recent studies reported associations between antibiotic use and breast cancer risk via effects on inflammation, immune function, and estrogen and phytochemical metabolism [9]. Friedman et al. reported that in 2.1 million women followed up for 9 years, the use of any antibiotic was related to a slightly increased risk of developing breast cancer [Hazard ratio = 1.14; 95% confidence interval: 1.10–1.18] [68]. However, Basso et al. reported that ansamycin may be a beneficial HER2-positive breast cancer treatment by inhibiting the “Akt dependent pathway” and cyclin D expression [69]. We observed that the “focal adhesion pathway” changed considerably and was enriched by ITGB1, ITGA4, and nine other up-regulated genes. Strelnikov et al. claimed a strong association between abnormal ITGA4 and ITGB1 hypermethylation and HER2-positive tumors [70]. Previous studies indicated that microenvironment-related pathways, such as “focal adhesion”, “ECM-receptor interaction”, and “complement and coagulation cascades” identified in this study are closely related to tumor initiation, disease progression, and metastasis, which are important future research directions [71, 72]. In addition, we identified significant changes in metabolism-related pathways, especially in the Luminal B group, such as “glycosaminoglycan biosynthesis”, “proteoglycans in cancer”, “tryptophan metabolism”, and “β-alanine metabolism”. We hypothesize these pathways are associated with dietary intake and improvements in living standards [73]; encouraging results from animal studies and clinical trials revealed the clinical relevance of these pathways and the benefit of targeted drugs for cancer [74,75,76,77]. Interestingly, Budczies et al. reported that β-alanine accumulated in breast cancer tissues, especially in the ER-negative subtype, in agreement with our results [78].
Notably, we observed eight up-regulated genes in “AMPK signaling” in HER2-enriched patients, whose activity may retard the growth of several cancers. Jhaveri et al. showed that AMPK regulated HER2 activity in HER2-enriched breast cancer cells, therefore AMPK activation may elicit a therapeutic benefit for such cancers [79, 80]. The “alcoholism pathway”, enriched in Luminal A, suggested an elevated risk for breast cancer. Recent evidence suggested that every alcohol unit/day enhanced the possibility of breast cancer by 7–11%, and this process was mechanistically underpinned by increased estrogen levels, acetaldehyde, and oxidative stress [81, 82]. Research also showed that disulfiram, an anti-alcoholism drug used in the clinic, induced apoptosis in vitro breast cancer cells and showed potential therapeutic candidacy [83]. Beyond that, hormone dependence is a concerning issue; menopause hormone therapy and plasticizers used in daily life are closely associated with ER-pathway activation, potentially contributing to breast cancer [84,85,86]. The HABITS trial reported that estrogen and progestogen doses may be associated with breast cancer recurrence [87].
In this study, we determined that genes and molecular pathways are constantly changing, suggesting molecular typing technologies must keep pace with this dynamic situation. Therefore, new biomarkers or pathways must be explored based on traditional molecular types. Our study had many limitations; small sample size and short period. Also, our analyses may not have fully reflected influences from the environment, time, habits, and other factors. Similarly, our study was an exploratory, retrospective analysis and lacked external validation using other methods. Thus, to some extent, the effectiveness and representation of the TCGA database are limited. Nonetheless, ours is the first study to investigate tumor genomic changes from a historical perspective. Although limited, our work provides new research directions and instills debate on this key issue. The observation of dynamic tumor genomic changes has the potential to support and reinforce existing cancer prevention strategies, drug development research programs, and prognostic predictions.
Materials and methods
Data sources
The Cancer Genome Atlas-Breast Cancer (TCGA-BRCA) RNAseqV2 gene expression and clinical data were acquired from the TCGA data portal (https://cancergenome.nih.gov) [88]. The “SummarizedExperiment” Bioconductor package (http://www.bioconductor.org) was used to complete and normalize data files in R (version 4.0.0, R Foundation for Statistical Computing). RNA-seq data from TCGA-BRCA covering 57,035 protein-coding and non-coding genes were used for analysis. We included 1102 patients diagnosed with breast cancer between 1988 and 2011; they were classified into eight groups according to the year of diagnosis. Patients diagnosed between 1988 and 1989 and 2000 and 2005 were classified as two separate groups to balance patient numbers in each group. Gene names were annotated to “Ensemble-id” according to corresponding TCGA platform files.
Gene identification
Expression changes of a particular gene in each group were defined as its average expression change in all breast cancers in that group. Genes whose expression levels were higher than those in the previous group, ≥ 6 times, were defined as up-regulated, and those whose expression levels were lower were defined as down-regulated. We produced heatmaps of the top 50 up-regulated and top 50 down-regulated genes identified in order of the log2 fold-change of the gene average expression of the last group (2011) to the initial group (1988-2000) using the “heatmap” package (version 2.7.7) in RStudio. Venn diagrams were also generated to identify up-regulated and down-regulated genes, known oncogenic genes, tumor suppressor genes (TSG) in the “OncoVar” database (https://oncovar.org), and known long intergenic non-coding RNA (lincRNA) in the “LNCipedia” database [89] using the “VennDiagram” package (version 1.6.20) in R.
Kyoto encyclopedia of genes and genomes pathway enrichment analysis
KEGG is a practical database that contains molecular information used to predict pathways where particular genes are enriched [90]. KEGG enrichment analyses were performed using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) (https://david.ncifcrf.gov). A P < 0.05 value was accepted as statistically significant. Breast cancers were classified into subtypes based on gene expression data. PAM50 breast cancer subtyping was widely used to classify breast cancer into four genotype groups: Luminal A, Luminal B, basal-like, and HER2-enriched [14]. We selected patients using the PAM50 subtypes from TCGA clinical information and the same analysis was performed for these four genotyping groups.
Statistical analyses
Patients with breast cancer were assigned to high and low expression groups based on the auto best cutoff of up-regulated or down-regulated gene expression levels as calculated by “survminer” (version 0.4.8) and “survival” (version 3.1) packages in RStudio. OS was the time from the date of diagnosis to the date of death due to any cause, or the last follow-up date. The survival probability of high and low expression groups was calculated using the Kaplan-Meier method and compared using log-rank tests. We also used line charts to display log2 expression trends in altered genes, and the proportion of high expression population of changing genes during eight periods. Altered genes not belonging to lincRNAs, Kaplan-Meier P values < 0.05, log2 fold-change in expression > 1 when comparing the last group (2011) with the initial group (1998-2000), and one drop allowed but the change of log2 expression less than one-third of the total change (2011 vs. 1988-2000), are shown (Figs. 3 and 4).
Conclusions
In conclusion, we analyzed changes in gene expression in breast cancer populations over two decades using the TCGA database. Our results proved that genes and molecular pathways are constantly changing, more importantly, some altered genes were associated with prognostics and are potential therapeutic targets. Our findings also suggest that the current molecular subtyping system of breast cancer should also be updated to keep pace with this dynamic situation.
Availability of data and materials
The original data used to support the findings of this study are available from TCGA Research Network (https://www.cancer.gov/tcga). The data sets and the R codes used in the current study are available from the corresponding author upon reasonable request.
References
Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021. https://doi.org/10.3322/caac.21660.
Malik A, Hafeez K, Nazar W, et al. Assessment of controversial risk factors in development of breast cancer: a study from local population. Biol Clin Sci Res J. 2021;1. https://doi.org/10.54112/bcsrj.v2021i1.49.
Schottenfeld D, Fraumeni JF, et al. Cancer epidemiology and prevention. 3rd ed: Oxford University Press; 2006.
Chlebowski RT, Manson JE, Anderson GL, et al. Estrogen plus progestin and breast cancer incidence and mortality in the Women’s health initiative observational study. J Natl Cancer Inst. 2013;105(8):526–35.
Althuis MD, Dozier JM, Anderson WF, Devesa SS, Brinton LA. Global trends in breast cancer incidence and mortality 1973–1997. Int J Epidemiol. 2005;34(2):405–12.
Colditz GA, Sellers TA, Trapido E. Epidemiology—identifying the causes and preventability of cancer? Nat Rev Cancer. 2006;6(1):75–83.
White AJ, O’Brien KM, Niehoff NM, Carroll R, Sandler DP. Metallic air pollutants and breast cancer risk in a nationwide cohort study. Epidemiology (Cambridge, Mass). 2019;30(1):20.
NTP (National Toxicology Program). Report on carcinogens, Fifteenth Edition. Research Triangle Park: U.S. Department of Health and Human Services, Public Health Service; 2021. https://ntp.niehs.nih.gov/go/roc15
Velicer CM, Heckbert SR, Lampe JW, Potter JD, Robertson CA, Taplin SH. Antibiotic use in relation to the risk of breast cancer. JAMA. 2004;291(7):827–35. https://doi.org/10.1001/jama.291.7.827.
Wang L. Early Diagnosis of Breast Cancer. Sensors (Basel). 2017;17(7). https://doi.org/10.3390/s17071572.
Nounou MI, ElAmrawy F, Ahmed N, Abdelraouf K, Goda S, Syed-Sha-Qhattal H. Breast Cancer: conventional diagnosis and treatment modalities and recent patents and technologies. Breast Cancer (Auckl). 2015;9(Suppl 2):17–34. https://doi.org/10.4137/BCBCR.S29420.
Harbeck N, Penault-Llorca F, Cortes J, et al. Breast cancer. Nat Rev Dis Primers. 2019;5(1):66. https://doi.org/10.1038/s41572-019-0111-2.
Sackett DL, Rosenberg WM, Gray JA, Haynes RB, Richardson WS. Evidence based medicine: what it is and what it isn't. 1996. Clin Orthop Relat Res. 2007;455:3–5.
Parker JS, Mullins M, Cheang MCU, et al. Supervised risk predictor of breast Cancer based on intrinsic subtypes. J Clin Oncol. 2009;27(8):1160–7. https://doi.org/10.1200/jco.2008.18.1370.
Cardoso F, van’t Veer LJ, Bogaerts J, et al. 70-gene signature as an aid to treatment decisions in early-stage breast cancer. N Engl J Med. 2016;375(8):717–29.
Dowsett M, Sestak I, Lopez-Knowles E, et al. Comparison of PAM50 risk of recurrence score with onco type DX and IHC4 for predicting risk of distant recurrence after endocrine therapy. J Clin Oncol. 2013;31(22):2783–90.
Toss A, Cristofanilli M. Molecular characterization and targeted therapeutic approaches in breast cancer. Breast Cancer Res. 2015;17(1):60. Published 2015 Apr 23. https://doi.org/10.1186/s13058-015-0560-9.
Alva AS, Mangat PK, Garrett-Mayer E, et al. Pembrolizumab in patients with metastatic breast Cancer with high tumor mutational burden: results from the targeted agent and profiling utilization registry (TAPUR) study. J Clin Oncol. 2021;39(22):2443–51. https://doi.org/10.1200/JCO.20.02923.
Gianni L, Eiermann W, Semiglazov V, et al. Neoadjuvant chemotherapy with trastuzumab followed by adjuvant trastuzumab versus neoadjuvant chemotherapy alone, in patients with HER2-positive locally advanced breast cancer (the NOAH trial): a randomised controlled superiority trial with a parallel HER2-negative cohort. Lancet. 2010;375(9712):377–84.
Schmid P, Rugo HS, Adams S, et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020;21(1):44–59. https://doi.org/10.1016/S1470-2045(19)30689-8.
Von Minckwitz G, Huang C-S, Mano MS, et al. Trastuzumab emtansine for residual invasive HER2-positive breast cancer. N Engl J Med. 2019;380(7):617–28.
Masuda N, Lee S-J, Ohtani S, et al. Adjuvant capecitabine for breast cancer after preoperative chemotherapy. N Engl J Med. 2017;376(22):2147–59.
Davies C, Pan H, Godwin J, et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet. 2013;381(9869):805–16.
World Health Organization. Global Health and Aging: World Health Organization; 2020. https://www.who.int/ageing/publications/global_health.pdf?ua=1. Accessed 01 Feb 2020
World Health Organization. Global health situation and projections estimates: World Health Organization; 1992.
Shuman EK. Global climate change and infectious diseases. N Engl J Med. 2010;362(12):1061–3. https://doi.org/10.1056/NEJMp0912931.
Casadevall A. Climate change brings the specter of new infectious diseases. J Clin Invest. 2020;130(2):553–5. https://doi.org/10.1172/JCI135003.
Cohen ML. Changing patterns of infectious disease. Nature. 2000;406(6797):762–7. https://doi.org/10.1038/35021206.
Smigal C, Jemal A, Ward E, et al. Trends in breast cancer by race and ethnicity: update 2006. CA Cancer J Clin. 2006;56(3):168–83.
van Der Meer DJ, Kramer I, van Maaren MC, et al. Comprehensive trends in incidence, treatment, survival, and mortality of first primary invasive breast cancer stratified by age, stage, and receptor subtype in the Netherlands between 1989 and 2017. Int J Cancer. 2021;148(9):2289–303.
DeSantis CE, Ma J, Gaudet MM, et al. Breast cancer statistics, 2019. CA Cancer J Clin. 2019;69(6):438–51.
Fisher B, Jeong J-H, Bryant J, et al. Treatment of lymph-node-negative, oestrogen-receptor-positive breast cancer: long-term findings from National Surgical Adjuvant Breast and bowel project randomised clinical trials. Lancet. 2004;364(9437):858–68.
Abe O, Abe R, Enomoto K, et al. Tamoxifen for early breast cancer: an overview of the randomised trials. Lancet. 1998;351(9114):1451–67.
Perez EA, Romond EH, Suman VJ, et al. Trastuzumab plus adjuvant chemotherapy for human epidermal growth factor receptor 2–positive breast cancer: planned joint analysis of overall survival from NSABP B-31 and NCCTG N9831. J Clin Oncol. 2014;32(33):3744.
Mavroudis D, Saloustros E, Malamos N, et al. Corrigendum to Six Versus 12 Months of Adjuvant Trastuzumab in Combination With Dose-Dense Chemotherapy for Women With HER2-positive Breast Cancer: A Multicenter Randomized Study by the Hellenic Oncology Research Group (HORG): Annals of Oncology, Volume 26, Issue 7, July 2015, Pages 1333-1340. Ann Oncol. 2020;31(3):444–5.
Mamounas EP, Tang G, Fisher B, et al. Association between the 21-gene recurrence score assay and risk of locoregional recurrence in node-negative, estrogen receptor–positive breast cancer: results from NSABP B-14 and NSABP B-20. J Clin Oncol. 2010;28(10):1677.
Van De Vijver MJ, He YD, Van't Veer LJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347(25):1999–2009.
Tian S, Roepman P, Van't Veer LJ, Bernards R, de Snoo F, Glas AM. Biological functions of the genes in the mammaprint breast cancer profile reflect the hallmarks of cancer. Biomark Insights. 2010;5:129–38. Published 2010 Nov 28. https://doi.org/10.4137/BMI.S6184.
Cristofanilli M, Turner NC, Bondarenko I, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016;17(4):425–39.
Bachelot T, Bourgier C, Cropet C, et al. Randomized phase II trial of everolimus in combination with tamoxifen in patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer with prior exposure to aromatase inhibitors: a GINECO study. J Clin Oncol. 2012;30(22):2718–24. https://doi.org/10.1200/JCO.2011.39.0708.
Baselga J, Im S-A, Iwata H, et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18(7):904–16.
Turner NC, Neven P, Loibl S, Andre F. Advances in the treatment of advanced oestrogen-receptor-positive breast cancer. Lancet. 2017;389(10087):2403–14.
Loibl S, Gianni L. HER2-positive breast cancer. Lancet. 2017;389(10087):2415–29.
Arteaga CL, Sliwkowski MX, Osborne CK, Perez EA, Puglisi F, Gianni L. Treatment of HER2-positive breast cancer: current status and future perspectives. Nat Rev Clin Oncol. 2012;9(1):16.
De Laurentiis M, Cianniello D, Caputo R, et al. Treatment of triple negative breast cancer (TNBC): current options and future perspectives. Cancer Treat Rev. 2010;36:S80–6.
Loibl S, Poortmans P, Morrow M, Denkert C, Curigliano G. Breast cancer. Lancet. 2021;397(10286):1750–69. https://doi.org/10.1016/S0140-6736(20)32381-3.
Robson M, Im S-A, Senkus E, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med. 2017;377(6):523–33.
Hoy SM. Talazoparib: first global approval. Drugs. 2018;78(18):1939–46.
Litton JK, Rugo HS, Ettl J, et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med. 2018;379(8):753–63.
Aressy B, Jullien D, Cazales M, et al. A screen for deubiquitinating enzymes involved in the G2/M checkpoint identifies USP50 as a regulator of HSP90-dependent Wee1 stability. Cell Cycle. 2010;9(18):3815–22. https://doi.org/10.4161/cc.9.18.13133.
Smith P, Nicholson LJ, Syed N, et al. Epigenetic inactivation implies independent functions for insulin-like growth factor binding protein (IGFBP)-related protein 1 and the related IGFBPL1 in inhibiting breast cancer phenotypes. Clin Cancer Res. 2007;13(14):4061–8. https://doi.org/10.1158/1078-0432.Ccr-06-3052.
Dai DN, Li Y, Chen B, et al. Elevated expression of CST1 promotes breast cancer progression and predicts a poor prognosis. J Mol Med (Berl). 2017;95(8):873–86. https://doi.org/10.1007/s00109-017-1537-1.
Sundvall M, Iljin K, Kilpinen S, Sara H, Kallioniemi OP, Elenius K. Role of ErbB4 in breast cancer. J Mammary Gland Biol Neoplasia. 2008;13(2):259–68. https://doi.org/10.1007/s10911-008-9079-3.
Bhan A, Soleimani M, Mandal SS. Long noncoding RNA and cancer: a new paradigm. Cancer Res. 2017;77(15):3965–81.
Yang G, Lu X, Yuan L. LncRNA: a link between RNA and cancer. Biochim Biophys Acta. 2014;1839(11):1097–109. https://doi.org/10.1016/j.bbagrm.2014.08.012.
Zhang T, Hu H, Yan G, et al. Long non-coding RNA and breast cancer. Technol Cancer Res Treat. 2019;18:1533033819843889.
Seroussi E, Kedra D, Pan HQ, et al. Duplications on human chromosome 22 reveal a novel ret finger protein-like gene family with sense and endogenous antisense transcripts. Genome Res Sep 1999;9(9):803-814. doi:https://doi.org/10.1101/gr.9.9.803.
Bu X, Qin A, Luo Z, Hu Y. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2019;44(7):741–8. https://doi.org/10.11817/j.issn.1672-7347.2019.190142.
Li H, Huang H, Li S, Mei H, Cao T, Lu Q. Long non-coding RNA ADAMTS9-AS2 inhibits liver cancer cell proliferation, migration, and invasion. Exp Ther Med. 2021;21(6):559. https://doi.org/10.3892/etm.2021.9991.
Zhang Z, Pi J, Zou D, et al. microRNA arm-imbalance in part from complementary targets mediated decay promotes gastric cancer progression. Nat Commun. 2019;10(1):4397. https://doi.org/10.1038/s41467-019-12292-5.
Xiong HG, Li H, Xiao Y, et al. Long noncoding RNA MYOSLID promotes invasion and metastasis by modulating the partial epithelial-mesenchymal transition program in head and neck squamous cell carcinoma. J Exp Clin Cancer Res. 2019;38(1):278. https://doi.org/10.1186/s13046-019-1254-4.
Han Y, Wu N, Jiang M, et al. Long non-coding RNA MYOSLID functions as a competing endogenous RNA to regulate MCL-1 expression by sponging miR-29c-3p in gastric cancer. Cell Prolif. 2019;52(6):e12678. https://doi.org/10.1111/cpr.12678.
Felsky D, Roostaei T, Nho K, et al. Neuropathological correlates and genetic architecture of microglial activation in elderly human brain. Nat Commun. 2019;10(1):409. https://doi.org/10.1038/s41467-018-08279-3.
Huang WJ, Tian XP, Bi SX, et al. The β-catenin/TCF-4-LINC01278-miR-1258-Smad2/3 axis promotes hepatocellular carcinoma metastasis. Oncogene. 2020;39(23):4538–50. https://doi.org/10.1038/s41388-020-1307-3.
Qu Z, Li S. Long noncoding RNA LINC01278 favors the progression of osteosarcoma via modulating miR-133a-3p/PTHR1 signaling. J Cell Physiol. 2020. https://doi.org/10.1002/jcp.29582.
Bi H, Shang Z, Jia C, et al. LncRNA RNF144A-AS1 promotes bladder Cancer progression via RNF144A-AS1/miR-455-5p/SOX11 Axis. Onco Targets Ther. 2020;13:11277–88. https://doi.org/10.2147/ott.S266067.
Huynh NP, Gloss CC, Lorentz J, et al. Long non-coding RNA GRASLND enhances chondrogenesis via suppression of the interferon type II signaling pathway. Elife. 2020;9. https://doi.org/10.7554/eLife.49558.
Friedman GD, Oestreicher N, Chan J, Quesenberry CP, Udaltsova N, Habel LA. Antibiotics and risk of breast cancer: up to 9 years of follow-up of 2.1 million women. Cancer epidemiology and prevention. Biomarkers. 2006;15(11):2102–6.
Basso AD, Solit DB, Munster PN, Rosen N. Ansamycin antibiotics inhibit Akt activation and cyclin D expression in breast cancer cells that overexpress HER2. Oncogene. 2002;21(8):1159–66.
Strelnikov VV, Kuznetsova EB, Tanas AS, et al. Abnormal promoter DNA hypermethylation of the integrin, nidogen, and dystroglycan genes in breast cancer. Sci Rep. 2021;11(1):2264. Published 2021 Jan 26. https://doi.org/10.1038/s41598-021-81851-y.
Luo M, Guan J-L. Focal adhesion kinase: a prominent determinant in breast cancer initiation, progression, and metastasis. Cancer Lett. 2010;289(2):127–39.
Soysal SD, Tzankov A, Muenst SE. Role of the tumor microenvironment in breast cancer. Pathobiology. 2015;82(3-4):142–52.
Koundouros N, Karali E, Tripp A, et al. Metabolic fingerprinting links oncogenic PIK3CA with enhanced Arachidonic acid-derived eicosanoids. Cell. 2020;181(7):1596–1611.e27. https://doi.org/10.1016/j.cell.2020.05.053.
Yip GW, Smollich M, Götte M. Therapeutic value of glycosaminoglycans in cancer. Mol Cancer Ther. 2006;5(9):2139–48.
Long J-P, Li X-N, Zhang F. Targeting metabolism in breast cancer: how far we can go? World J Clin Oncol. 2016;7(1):122.
Iozzo RV, Sanderson RD. Proteoglycans in cancer biology, tumour microenvironment and angiogenesis. J Cell Mol Med. 2011;15(5):1013–31.
Juhász C, Nahleh Z, Zitron I, et al. Tryptophan metabolism in breast cancers: molecular imaging and immunohistochemistry studies. Nucl Med Biol. 2012;39(7):926–32.
Budczies J, Brockmöller SF, Müller BM, et al. Comparative metabolomics of estrogen receptor positive and estrogen receptor negative breast cancer: alterations in glutamine and beta-alanine metabolism. J Proteome. 2013;94:279–88.
Hadad SM, Fleming S, Thompson AM. Targeting AMPK: a new therapeutic opportunity in breast cancer. Crit Rev Oncol Hematol. 2008;67(1):1–7. https://doi.org/10.1016/j.critrevonc.2008.01.007.
Jhaveri TZ, Woo J, Shang X, Park BH, Gabrielson E. AMP-activated kinase (AMPK) regulates activity of HER2 and EGFR in breast cancer. Oncotarget. 2015;6(17):14754–65. https://doi.org/10.18632/oncotarget.4474.
Janbaz KH, Qadir MI, Siddiq Z. Stages, alcoholism and genetic basis of breast cancer. Acad Res Int. 2011;1(2):383.
Seitz HK, Pelucchi C, Bagnardi V, Vecchia CL. Epidemiology and pathophysiology of alcohol and breast cancer: update 2012. Alcohol Alcohol. 2012;47(3):204–12.
Chen D, Cui QC, Yang H, Dou QP. Disulfiram, a clinically used anti-alcoholism drug and copper-binding agent, induces apoptotic cell death in breast cancer cultures and xenografts via inhibition of the proteasome activity. Cancer Res. 2006;66(21):10425–33.
López-Carrillo L, Hernández-Ramírez RU, Calafat AM, et al. Exposure to phthalates and breast cancer risk in northern Mexico. Environ Health Perspect. 2010;118(4):539–44.
Jobling S, Reynolds T, White R, Parker MG, Sumpter JP. A variety of environmentally persistent chemicals, including some phthalate plasticizers, are weakly estrogenic. Environ Health Perspect. 1995;103(6):582–7.
Santen RJ. Menopausal hormone therapy and breast cancer. J Steroid Biochem Mol Biol. 2014;142:52–61. https://doi.org/10.1016/j.jsbmb.2013.06.010.
von Schoultz E, Rutqvist LE. On behalf of the Stockholm breast Cancer study G. menopausal hormone therapy after breast Cancer: the Stockholm randomized trial. J Natl Cancer Inst. 2005;97(7):533–5. https://doi.org/10.1093/jnci/dji071.
Cancer Genome Atlas Research N, Weinstein JN, Collisson EA, et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45(10):1113–20. https://doi.org/10.1038/ng.2764.
Volders PJ, Anckaert J, Verheggen K, et al. LNCipedia 5: towards a reference set of human long non-coding RNAs. Nucleic Acids Res. 2019;47(D1):D135–d139. https://doi.org/10.1093/nar/gky1031.
Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45(D1):D353–61. https://doi.org/10.1093/nar/gkw1092.
Acknowledgments
All data used in this publication were generated by the TCGA project. The authors declare no competing interest.
Funding
This work was supported by the National Key Research and Development Program of China (Grant No. 2021YFE0203200), the National Natural Science Foundation of China (Grant No. 92059105), the Beijing Municipal Natural Science Foundation (Grant No. 7202212), the Research and Development Funds of Peking University People’s Hospital (Grant Nos. RDX2021-05, RDY2020-16) and the Young Investigator Program of Peking University Health Science Center (Grant No. BMU2021PYB013).
Author information
Authors and Affiliations
Contributions
Shu Wang contributed to the study design, Jinbo Wu, Hongjun Liu and Taobo Hu contributed to data analysis, and all authors contributed to manuscript writing. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval and consent to participate
Patient data were obtained from The Cancer Genome Atlas (TCGA) database, a publicly open database resource. Informed patient consent was not required.
Consent for publication
Not applicable.
Competing interests
The authors declare none.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wu, J., Liu, H., Hu, T. et al. Gene expression trend changes in breast cancer populations over two decades: insights from The Cancer Genome Atlas database. Hereditas 159, 18 (2022). https://doi.org/10.1186/s41065-022-00230-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41065-022-00230-3