
Abstract
Background
A chronic progressive degenerative joint disease, such as osteoarthritis (OA) is positively related to age. The medical economy is facing a major burden, because of the high disability rate seen in patients with OA. Therefore, to prevent and treat OA, exploring the diagnostic biomarkers of OA will be of great significance.
Methods
Differentially expressed genes (DEGs) were obtained from the Gene Expression Omnibus database using the RobustRankAggreg R package, and a protein–protein interaction network was constructed. The module was obtained from Cytoscape, and the four algorithms of degree, MNC, closeness, and MCC in CytoHubba were used to identify the hub genes. A diagnostic model was constructed using Support Vector Machines (SVM), and the ability of the model to predict was evaluated by other cohorts.
Results
From normal and OA samples, 136 DEGs were identified, out of which 45 were downregulated in the normal group and 91 were upregulated in the OA group. These genes were associated with the extracellular matrix-receptor interactions, the PI3K-Akt signaling pathway, and the protein digestion and absorption pathway, as per a functional enrichment analysis. Finally, we identified the 7 hub genes (COL6A3, COL1A2, COL1A1, MMP2, COL3A1, POST, and FN1). These genes have important roles and are widely involved in the immune response, apoptosis, inflammation, and bone development. These 7 genes were used to construct a diagnostic model by SVM, and it performed well in different cohorts. Additionally, we verified the methylation expression of these hub genes.
Conclusions
The 7-genes signature can be used for the diagnosis of OA and can provide new ideas in the clinical decision-making for patients with OA.
Similar content being viewed by others


Introduction
Worldwide, the most common musculoskeletal disease osteoarthritis (OA) is seen in people aged 60 years and above. Recently, because of the increase in life expectancy, the vigorous development of sports, and the increase in the number of people with obesity, the number of young patients with OA is significantly increasing [1]. In the United States, OA has become a major cause of disability, resulting in huge social and economic burdens [2, 3]. A sensitive diagnosis of OA can delay the progression of the disease and can improve the prognoses of patients [4]. Presently, OA is diagnosed mainly by joint aspiration, X-ray, and magnetic resonance imaging (MRI) [5,6,7]. However, the local cartilage damage cannot be detected by joint aspiration and X-ray before any structural damage occurs [8, 9]. Although MRI is sensitive, it still cannot detect the localized degeneration of the cartilage tissue. Moreover, degeneration of the cartilage tissue is usually related to early pathogenesis of OA [10, 11]. Therefore, there are still certain limitations. A few researchers have proposed that the diagnosis can be done using optical probes based on hyaluronic acid [12], but whether they can be used in clinical practice is not yet confirmed.
xploring the sensitive and specific diagnostic biomarkers of OA is of great significance. In this study, we conducted a comprehensive analysis using multiple public microarray datasets (expression profiles and methylation data) to determine the potential transcriptome biomarkers of OA. Further, we established the gene regulatory networks based on the protein–protein interaction (PPI) network involved in these DEGs to identify the diagnostic biomarkers. Based on the Support Vector Machine (SVM) model, we have identified and established a diagnostic model for patients with OA.
Methods and materials
Data sources and downloads
We downloaded the chip expression dataset of OA (GSE129147 [13], GSE57218 [14], GSE51588 [15], and GSE117999 [16]) and the methylation dataset (GSE73626 [17]) from the Gene Expression Omnibus (GEO) database. We selected the chip dataset containing normal samples and OA samples.
Preprocessing of the data
The GEO datasets were processed as follows: 1) the normal and the OA samples were retained, 2) the probe was converted into a gene symbol, 3) the probe corresponding to multiple genes was removed, and 4) the median of the multiple gene symbol expressions were obtained. In the preprocessed datasets, GSE129147 had 9 normal and 10 OA samples, GSE57218 had 7 normal and 33 OA samples, GSE51588 had 10 normal and 40 OA samples, GSE117999 had 10 normal and 10 OA samples, and GSE73626 had 7 normal and 11 OA samples. The clinical statistics of these samples can be found in Table 1.
Identification of differentially expressed gene and functional analysis
Limma R package was used to calculate the differentially expressed genes (DEGs) between the normal and the OA samples of the GSE129147, GSE57218, and GSE51588 datasets. The DEGs were filtered according to the threshold FDR < 0.05 and |FC|> 1.5. A volcano map was drawn after obtaining the DEGs from the three datasets. The DEGs in the three datasets were analyzed using the RobustRankAggreg [18] R package. According to the score, the heat maps were drawn, and the DEGs were retained. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis was used to analyze the DEGs in the OA samples, and WebGestaltR (v0.4.2) R package was used to analyze the Gene Ontology (GO) functional enrichment analysis.
MCODE module screening and functional analysis
To study the interaction network between proteins and help mine core regulatory genes, the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database (https://string-db.org/) can be used. The STRING database was used to analyze the PPI network of the DEGs. The resulting files were screened using Cytoscape (v3.7.2), and the network module was obtained using the MCODE plug-in algorithm. The KEGG pathway analysis and the GO functional enrichment analysis were performed on the genes in the MCODE module using the WebGestaltR (v0.4.2) R package.
Identification of the hub genes
The four algorithms of degree, MNC, closeness, and MCC of the cytoHubba plug-in in Cytoscape (v3.7.2) were used to calculate the PPI network constructed by the DEGs, and the top 10 genes were selected as the hub genes. The hub genes obtained by these four algorithms were intersected with the genes of the functional module MCODE. A Wayne diagram was drawn after the final hub genes were obtained.
Construction and validation of the diagnostic model
The GSE51588 dataset was used as the training cohort, and the GSE57218, GSE129147, and GSE117999 datasets were used as the validation cohort. The hub genes were used as a feature in the training cohort to obtain the corresponding expression profiles; to build an SVM classification model; to calculate the classification accuracy, sensitivity, and specificity of the model; and the area under the receiver operating characteristic (ROC) curve.
Results
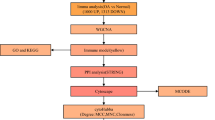
Flowchart
To analyze the hub genes and the diagnostic model of OA, we designed a flowchart (Fig. 1).

Flowchart displaying the characteristic genes and the diagnostic model of osteoarthritis
Identification of 136 DEGs
The DEGs between the normal and the OA samples of the GSE129147, GSE57218, and GSE51588 datasets were calculated using the limma R package.
The results showed 268 DEGs in the GSE129147 dataset, out of which 188 were upregulated and 80 were downregulated (S1_Table). There were 648 DEGs in the GSE57218 dataset, out of which 309 were upregulated and 339 were downregulated (S2_Table). There were 4297 DEGs in the GSE51588 dataset, out of which 2664 were upregulated and 1633 were downregulated (S3_Table). The volcano maps of the upregulated and the downregulated DEGs in the normal and the OA samples of the three datasets are shown in Fig. 2A–2C. Further, integration and analysis of the DEGs in the three datasets showed that 136 DEGs were obtained, out of which 91 were upregulated in the OA group (S4_Table) and 45 were downregulated in the normal group (S5_Table). The heat maps of the 136 DEGs are shown in Fig. 2D–2F. The heat map of the top 50 genes that were significantly upregulated and downregulated in each dataset is shown in Fig. 2G.

A: A volcano map of the differential genes in the GSE129147 dataset. B: A volcano map of the differential genes in the GSE57218 dataset. C: A volcano map of the differential genes in the GSE51588 dataset. D: A heat map of the differential genes in the GSE129147 dataset. E: A heat map of the differential genes in the GSE57218 dataset. F: A heat map of the differential genes in the GSE51588 dataset. G: A heat map of the partial differential genes in the three datasets
Functional analysis of the DEGs
The KEGG pathway analysis and the GO functional enrichment analysis were performed on the 136 DEGs. From the GO annotations of the DEGs in the OA samples, 211 items had significant biological process (BP) differences. The first 20 enriched annotations are shown in Fig. 3A. Humoral immune response, cartilage development involved in endochondral bone morphogenesis, bone morphogenesis, chondrocyte development involved in endochondral bone morphogenesis, endochondral bone morphogenesis, and other BP were enriched. Thirty-six items with significant molecular function (MF) differences were enriched, and the first 20 annotations are shown in Fig. 3C. There were significant differences in the 38 cellular components (CC) annotations (P < 0.05). The first 20 are shown in Fig. 3B. For the KEGG pathway enrichment in the OA samples, 36 items were annotated significantly. The first 20 are shown in Fig. 3D. Extracellular matrix (ECM)-receptor interactions, PI3K-Akt signaling pathway, and protein digestion and absorption were significantly enriched.

A: A biological process annotated map of the differential genes. B: A cellular component annotated map of differential genes. C: A molecular function annotated map of the differential genes. D: A KEGG annotated map of the differential genes
Identification and functional analysis of the MCODE module
The 136 DEGs in the PPI network were analyzed using the STRING database and were visualized using Cytoscape (v3.7.2). MCODE1 was found using the MCODE algorithm (Fig. 4). Furthermore, 27 genes in the MCODE1 module were analyzed by the pathway and the functional enrichment analysis. For the GO functional analysis, the first 20 annotations in BP are shown in Fig. 5A. Cartilage development involved in endochondral bone morphogenesis, cartilage development, bone morphogenesis, and bone development were significantly enriched. Seventeen annotations in MF were enriched (Fig. 5C). Nine annotations in CC were enriched (P < 0.05) (Fig. 5B). For the KEGG pathway enrichment of the MCODE module, nine significant annotations were enriched (Fig. 5D). The ECM-receptor interactions, the PI3K-Akt signaling pathway, the protein digestion and absorption, and the focal adhesion pathway were significantly enriched.

A gene protein–protein interaction map of functional modules mined by MCODE

A: A biological process annotation map of the MCODE module genes. B: A cellular component annotation map of the MCODE module genes. C: A molecular function annotation map of the MCODE module genes. D: A KEGG annotation map of the MCODE module genes
Construction and validation of the 7-genes diagnostic model
We used the four algorithms of degree, MNC, closeness, and MCC of cytoHubba to calculate the 136 DEGs in the PPI network, and then selected the top 10 genes as the hub genes. The PPI network of these hub genes is shown in Fig. 6. The hub genes obtained by these four algorithms were intersected with the genes in the MCODE module, and thus, the 7 hub genes, including COL6A3, COL1A2, COL1A1, MMP2, COL3A1, POSTN, and FN1 were obtained (Fig. 7). Additionally, we compared the expression of these 7 hub genes in the normal and the OA samples with other datasets (GSE129147, GSE57218, and GSE51588), and the results showed that all the 7 genes were highly expressed in the OA samples (Fig. 8).

A: A protein–protein interaction (PPI) network diagram of the hub genes using the degree algorithm. B: A PPI network diagram of the hub genes using the MCC algorithm. C: A PPI network diagram of the hub genes using the MNC algorithm. D: A PPI network diagram of the hub genes using the closeness algorithm

Wayne diagram displaying the hub genes

A: Expression of the hub genes in the GSE129147 dataset. B: Expression of the hub genes in the GSE57218 dataset. C: Expression of the hub genes in the GSE51588 dataset
The SVM classification model was constructed using the 7 hub genes as a feature in the training cohort. A 100% classification accuracy rate was obtained. Of the 50 samples, all 50 were correctly classified, the sensitivity and specificity of the model were both 100%, and the area under the ROC curve (AUC) was 1 (Fig. 9A). The GSE57218 dataset was used for verification. Thirty-nine of the 40 samples were correctly classified; the classification accuracy rate was 97.5%, the sensitivity of the model was 100%, the specificity was 85.7%, and the AUC was 0.93 (Fig. 9B). The GSE129147 dataset was used for verification. Eighteen of the 19 samples were correctly classified; the classification accuracy rate was 94.7%, the sensitivity of the model was 90%, the specificity was 100%, and the AUC was 0.95 (Fig. 9C). Finally, the GSE117999 dataset was used for verification. Twenty of the 20 samples were correctly classified; the classification accuracy rate was 100%, the sensitivity of the model was 100%, the specificity was 100%, and the AUC was 1 (Fig. 9D). These results show that the diagnostic prediction model constructed in this study could effectively distinguish between the normal and the OA samples, and the 7 hub genes could be used as reliable biomarkers for diagnosing OA.

A: Classification result and receiver operating characteristic (ROC) curve of the diagnostic model in the GSE51588 dataset. B: Classification result and ROC curve of the diagnostic model in the GSE57218 dataset. C: Classification result and ROC curve of the diagnostic model in the GSE129147 dataset. D: Classification result and ROC curve of the diagnostic model in the GSE117999 dataset
Methylation analysis of the hub genes
The GSE73626 dataset was used to analyze the methylation of the 7 hub genes in the normal and the OA samples. Information on the annotation of the methylation sites and the distance between the probe CpG site and the transcription start site is compiled in S6_Table.The results showed that the methylation sites of COL6A3, COL1A2, COL1A1, MMP2, COL3A1, POSTN, and FN1 were methylated in the normal samples. The normal samples had higher overall methylation than the OA samples, which is consistent with the fact that the expression of these 7 genes in the normal samples was lower than that in the OA samples. Fig. 10Methylation was negatively correlated with gene expression.

Methylation sites of a few of the hub genes undergoing methylation
Discussion
OA is a chronic progressive degenerative joint disease having an incidence positively correlated with age. Joint pain, swelling, and limited mobility are the main clinical manifestations of OA. The current treatments are mainly aimed at alleviating joint pain and reducing joint disability, which has become a major burden on the medical economy. In 1986, Poulter et al. proposed that immunocytological examination of the synovial fluid might have a diagnostic and prognostic value for OA [19]. However, so far, the pathogenesis of OA is still unclear. Investigating the pathogenesis of OA in-depth and exploring the biomarkers for diagnosing OA have important health and economic benefits.
In this study, we identified DEGs between the normal and the OA samples in three datasets (GSE129147, GSE57218, and GSE51588) using the RobustRankAggreg R package, and 136 DEGs were identified. A PPI network was constructed based on these DEGs, and MCODE1 was selected. The genes in this module were mainly involved in endochondral bone morphogenesis, cartilage development, and bone morphogenesis.
Endochondral bone morphogenesis results from chondrocyte differentiation, hypertrophy, death, and bone replacement. This process is extremely important for normal bone growth and development and fracture repair. Endochondral bone formation can be promoted by inhibiting apoptosis signal-regulated kinase 1 and by increasing the survival rate of chondrocytes, thereby slowing the progression of OA [20]. Pathway analysis showed that the ECM-receptor interactions, the PI3K-Akt signaling pathway, the protein digestion and absorption, and the focal adhesion pathway were significantly enriched.
Studies have shown that the above three signaling pathways play an important role in the occurrence and development of OA. Among them, ASG-IV might regulate the Hippo signaling pathway by upregulating VTN and collagen type I alpha 1 (COL1A1) involved in the ECM-receptor interactions, thus playing an important role in chondrocyte apoptosis in human OA [21]. Fibroblast growth factor 18 exerts an anti-OA effect by fusing and dividing the PI3K-Akt signaling pathway and the mitochondria [22]. Downregulation of microRNA 34a (MiR-34a) promotes chondrocyte proliferation and inhibits apoptosis by activating the PI3K-Akt signaling pathway in chondrocytes in rat OA [23]. Resveratrol inhibits the development of obesity-related OA through toll-like receptor 4 (TLR4) and the PI3K-Akt signaling pathway [24]. OA can be reduced in mice by deleting the focal adhesion mechanosensitive connector, hydrogen peroxide-inducible clone-5 (Hic-5) [25].
We used the four algorithms of Degree, MNC, Closeness, and MCC of the CytoHubba to calculate the 136 DEGs in the PPI network, and then selected the top 10 genes as the hub genes. The hub genes obtained using these four algorithms were intersected with the genes in MCODE1, and thus the 7 hub genes, including COL6A3, COL1A2, COL1A1, MMP2, COL3A1, POSTN, and FN1, were obtained. These genes are widely involved in immune response, apoptosis, inflammation, and bone development. Collagen type VI-related myopathies are inherited myopathies characterized by muscle weakness, atrophy, and joint contracture as the main clinical manifestations, because of the functional defects in the type VI collagen [26]. The α1, α2, and α3 peptide chains are composed of heterologous peptide chain monomers through triple-helix structures. The active type VI collagen is formed when the monomers form tetramers. These three peptide chains are encoded by the COL6A1, COL6A2, and COL6A3 genes, respectively [26, 27]. Collagen-related myopathies occur when any of these genes have pathogenic variants. COL1A1 is closely related to Caffey disease and type I osteogenesis imperfecta, and its related pathways include the integrin pathway and collagen chain trimerization [28,29,30,31,32]. Collagen type III alpha 1 (COL3A1) exists in the skin, lungs, intestinal walls, and blood vessel walls, and it can strengthen and support many tissues in the body [33,34,35]. Multicentric osteolysis, nodulosis, and arthropathy are caused by at least eight mutations in the matrix metalloproteinase-2 (MMP2) gene, which is a rare inherited bone disease characterized by loss of bone tissue (osteolysis), especially in the hands and feet. Each known MMP2 gene mutation eliminates the function of the MMP2 and prevents the normal cutting of type IV collagen. During bone modeling, losing enzyme activity might disrupt the balance between new bone formation and rupture of existing bones, leading to the gradual loss of bone tissue [36,37,38]. Periostin (POSTN) can promote accumulation and differentiation of the osteoblasts and their precursor cells in the periosteum [39], the damaged blood vessel reconstruction [40], and the proliferation and fibrosis of inflamed tissues [41]. In vivo and in vitro experiments have shown that adding a certain concentration of POSTN can increase the proliferation rate of knee tibial plateau chondrocytes. Intra-articular injection of a certain amount of PSTN may lead to new ideas to treat knee OA in the middle and the late stages. [42]. In terms of synovial chondropathy cases, 57% show Fibronectin-1 (FN1) and/or activin receptor type-2A [43]. The FN1-EGF gene fusion is common in calcified aponeurotic fibroma [44]. Spinal hypoplasia, some corner fractures, and glomerulopathy with fibronectin deposits are diseases associated with FN1 [45, 46]. Combined with the existing research, it can be speculated that the above 7 genes play an important role in the occurrence and development of OA, but their specific mechanisms still need to be explored.
We used these seven genes to construct a diagnostic model using the SVM method. The model displayed good performance in different datasets. Additionally, we verified the expression of a few hub genes, and the methylation was negatively correlated with gene expression, showing that to a certain extent, the expression of these genes is related to methylation regulation.
There are a few limitations to our study. The sample size in this study was limited, and the cohort was not large enough, which might have affected the statistical validity and accuracy of our results. Moreover, this study is based only on bioinformatic analysis. Therefore, complementary and basic experiments are still required to reveal the specific mechanism of action of the signature gene markers in the progression of OA. Further studies will be necessary to explore the underlying molecular mechanism of action of these genes to demonstrate their applicability in clinical applications.
In conclusion, we have systematically analyzed the expression of multiple GEO cohorts and found the expression characteristics of the 7 hub genes, according to the PPI network. These genes are associated with the occurrence and development of OA and have a high accuracy in predicting OA (AUC > 0.93), which provides a theoretical basis for diagnosing OA by clinicians .
Availability of data and materials
The data sets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- OA:
-
Osteoarthritis
- DEGs:
-
Differentially expressed genes
- SVM:
-
Support Vector Machines
- MRI:
-
Magnetic resonance imaging
- PPI:
-
Protein–protein interaction
- BP:
-
Biological process
- AUC:
-
Area under the ROC curve
- ASK1:
-
Apoptosis signal-regulated kinase 1
- COL1A1:
-
Collagen type I alpha 1
- FGF18:
-
Fibroblast growth factor 18
- TLR4:
-
Toll-like receptor 4
References
Sutton PM, Holloway ES. The young osteoarthritic knee: dilemmas in management. BMC Med. 2013;11:14.
Kotlarz H, et al. Insurer and out-of-pocket costs of osteoarthritis in the US: evidence from national survey data. Arthritis Rheum. 2009;60(12):3546–53.
Luo X, et al. Estimates and patterns of direct health care expenditures among individuals with back pain in the United States. Spine Phila Pa 1976. 2004;29(1):79–86.
Chu CR, et al. Early diagnosis to enable early treatment of pre-osteoarthritis. Arthritis Res Ther. 2012;14(3):212.
Eckstein F, Wirth W, Nevitt MC. Recent advances in osteoarthritis imaging–the osteoarthritis initiative. Nat Rev Rheumatol. 2012;8(10):622–30.
Ding C, Zhang Y, Hunter D. Use of imaging techniques to predict progression in osteoarthritis. Curr Opin Rheumatol. 2013;25(1):127–35.
Fernandez-Madrid F, et al. Synovial thickening detected by MR imaging in osteoarthritis of the knee confirmed by biopsy as synovitis. Magn Reson Imaging. 1995;13(2):177–83.
Guermazi A, et al. Why radiography should no longer be considered a surrogate outcome measure for longitudinal assessment of cartilage in knee osteoarthritis. Arthritis Res Ther. 2011;13(6):247.
Kraus VB. Waiting for action on the osteoarthritis front. Curr Drug Targets. 2010;11(5):518–20.
Kandahari AM, et al. Recognition of Immune Response for the Early Diagnosis and Treatment of Osteoarthritis. J Immunol Res. 2015;2015:192415.
de Lange-Brokaar BJ, et al. Synovial inflammation, immune cells and their cytokines in osteoarthritis: a review. Osteoarthritis Cartilage. 2012;20(12):1484–99.
Li S, et al. Hyaluronic Acid-Based Optical Probe for the Diagnosis of Human Osteoarthritic Cartilage. Nanotheranostics. 2018;2(4):347–59.
Asik MD, et al. Microarray analysis of cartilage: comparison between damaged and non-weight-bearing healthy cartilage. Connect Tissue Res. 2020;61(5):456–64.
Ramos YF, et al. Genes involved in the osteoarthritis process identified through genome wide expression analysis in articular cartilage; the RAAK study. PLoS One. 2014;9(7):e103056.
Chou CH, et al. Genome-wide expression profiles of subchondral bone in osteoarthritis. Arthritis Res Ther. 2013;15(6):R190.
Brophy RH, et al. Transcriptome comparison of meniscus from patients with and without osteoarthritis. Osteoarthritis Cartilage. 2018;26(3):422–32.
Aref-Eshghi E, et al. Genome-wide DNA methylation study of hip and knee cartilage reveals embryonic organ and skeletal system morphogenesis as major pathways involved in osteoarthritis. BMC Musculoskelet Disord. 2015;16:287.
Kolde R, et al. Robust rank aggregation for gene list integration and meta-analysis. Bioinformatics. 2012;28(4):573–80.
Poulter LW, et al. Immunocytology of synovial fluid cells may be of diagnostic and prognostic value in arthritis. Ann Rheum Dis. 1986;45(7):584–90.
Eaton GJ, et al. Inhibition of apoptosis signal-regulating kinase 1 enhances endochondral bone formation by increasing chondrocyte survival. Cell Death Dis. 2014;5:e1522.
Castelletti F, et al. Mutations in FN1 cause glomerulopathy with fibronectin deposits. Proc Natl Acad Sci U S A. 2008;105(7):2538–43.
Yao X, et al. Fibroblast growth factor 18 exerts anti-osteoarthritic effects through PI3K-AKT signaling and mitochondrial fusion and fission. Pharmacol Res. 2019;139:314–24.
Tao H, Cheng L, Yang R. Downregulation of miR-34a Promotes Proliferation and Inhibits Apoptosis of Rat Osteoarthritic Cartilage Cells by Activating PI3K/Akt Pathway. Clin Interv Aging. 2020;15:373–85.
Xu X, et al. Resveratrol inhibits the development of obesity-related osteoarthritis via the TLR4 and PI3K/Akt signaling pathways. Connect Tissue Res. 2019;60(6):571–82.
Miyauchi A, et al. Alleviation of murine osteoarthritis by deletion of the focal adhesion mechanosensitive adapter, Hic-5. Sci Rep. 2019;9(1):15770.
Lampe AK, Bushby KM. Collagen VI related muscle disorders. J Med Genet. 2005;42(9):673–85.
Allamand V, et al. 166th ENMC International Workshop on Collagen type VI-related Myopathies, 22–24 May 2009, Naarden. The Netherlands Neuromuscul Disord. 2010;20(5):346–54.
Jin H, et al. Polymorphisms in the 5’ flank of COL1A1 gene and osteoporosis: meta-analysis of published studies. Osteoporos Int. 2011;22(3):911–21.
Mann V, Ralston SH. Meta-analysis of COL1A1 Sp1 polymorphism in relation to bone mineral density and osteoporotic fracture. Bone. 2003;32(6):711–7.
Lim J, et al. Genetic causes and mechanisms of Osteogenesis Imperfecta. Bone. 2017;102:40–9.
Van Dijk FS, Sillence DO. Osteogenesis imperfecta: clinical diagnosis, nomenclature and severity assessment. Am J Med Genet A. 2014;164A(6):1470–81.
Schwarze U, et al. Rare autosomal recessive cardiac valvular form of Ehlers-Danlos syndrome results from mutations in the COL1A2 gene that activate the nonsense-mediated RNA decay pathway. Am J Hum Genet. 2004;74(5):917–30.
Beridze N, Frishman WH. Vascular Ehlers-Danlos syndrome: pathophysiology, diagnosis, and prevention and treatment of its complications. Cardiol Rev. 2012;20(1):4–7.
Malfait F, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175(1):8–26.
Oderich GS, et al. The spectrum, management and clinical outcome of Ehlers-Danlos syndrome type IV: a 30-year experience. J Vasc Surg. 2005;42(1):98–106.
Castberg FC, et al. Multicentric osteolysis with nodulosis and arthropathy (MONA) with cardiac malformation, mimicking polyarticular juvenile idiopathic arthritis: case report and literature review. Eur J Pediatr. 2013;172(12):1657–63.
Mosig RA, et al. Loss of MMP-2 disrupts skeletal and craniofacial development and results in decreased bone mineralization, joint erosion and defects in osteoblast and osteoclast growth. Hum Mol Genet. 2007;16(9):1113–23.
Tuysuz B, et al. A novel matrix metalloproteinase 2 (MMP2) terminal hemopexin domain mutation in a family with multicentric osteolysis with nodulosis and arthritis with cardiac defects. Eur J Hum Genet. 2009;17(5):565–72.
Litvin J, et al. Expression and function of periostin-isoforms in bone. J Cell Biochem. 2004;92(5):1044–61.
Li P, et al. Hypoxia-responsive growth factors upregulate periostin and osteopontin expression via distinct signaling pathways in rat pulmonary arterial smooth muscle cells. J Appl Physiol (1985). 2004;97(4):1550–8 (discussion 1549).
Jia G, et al. Periostin is a systemic biomarker of eosinophilic airway inflammation in asthmatic patients. J Allergy Clin Immunol. 2012;130(3):647–54. https://doi.org/10.1016/j.jaci.2012.06.025.
Gillan L, et al. Periostin secreted by epithelial ovarian carcinoma is a ligand for alpha(V)beta(3) and alpha(V)beta(5) integrins and promotes cell motility. Cancer Res. 2002;62(18):5358–64.
Amary F, et al. Synovial chondromatosis and soft tissue chondroma: extraosseous cartilaginous tumor defined by FN1 gene rearrangement. Mod Pathol. 2019;32(12):1762–71.
Puls F, et al. FN1-EGF gene fusions are recurrent in calcifying aponeurotic fibroma. J Pathol. 2016;238(4):502–7.
Hobeika L, et al. Characterization of glomerular extracellular matrix by proteomic analysis of laser-captured microdissected glomeruli. Kidney Int. 2017;91(2):501–11.
To WS, Midwood KS. Plasma and cellular fibronectin: distinct and independent functions during tissue repair. Fibrogenesis Tissue Repair. 2011;4:21.
Acknowledgements
Not applicable
Funding
Not applicable
Author information
Authors and Affiliations
Contributions
Yaguang and Han Jun Wu designed the current study. Jun Wu and Zhenyu Gong collected the data. Yaguang Han and Yiqin Zhou analyzed and interpreted the data. Jun Wu, Yaguang Han and Haobo Li wrote the manuscript. Yi Chen and Qirong Qian supervised the study. All authors read and approved the final version of the manuscript and agreed to be accountable for all aspects of the research in ensuring that the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Tableshowing the 268 differentially expressed genes in the GSE129147 dataset, ofwhich 188 were upregulated and 80 were downregulated
Additional file 2.
Tableshowing the 648 differentially expressed genes in the GSE57218 dataset, ofwhich 309 were upregulated and 339 were downregulated
Additional file 3.
Tableshowing the 4297 differentially expressed genes in the GSE51588 dataset, ofwhich 2664 were upregulated and 1633 were downregulated
Additional file 4.
Tableshowing the integration and analysis of the differentially expressed genes inthe three datasets, 136 differentially expressed genes were obtained, of which91 were upregulated in the OA group
Additional file 5.
Tableshowing that in the three datasets, 136 differentially expressed genes wereobtained, of which 45 were downregulated in the normal group
Additional file 6.
Informationon the annotation of the methylation sites and the distance between the probeCpG site and the transcription start site
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Han, Y., Wu, J., Gong, Z. et al. Identification and development of the novel 7-genes diagnostic signature by integrating multi cohorts based on osteoarthritis. Hereditas 159, 10 (2022). https://doi.org/10.1186/s41065-022-00226-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41065-022-00226-z