
Abstract
Background
The aim of this study was to identify the candidate biomarkers and pathways associated with psoriasis. GSE13355 and GSE14905 were extracted from the Gene Expression Omnibus (GEO) database. Then the differentially expressed genes (DEGs) with |logFC| > 2 and adjusted P < 0.05 were chosen. In addition, the Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses for DEGs were performed. Then, the GO terms with P < 0.05 and overlap coefficient greater than 0.5 were integrated by EnrichmentMap. Additionally, risk subpathways analysis for DEGs was also conducted by using the iSubpathwayMiner package to obtain more psoriasis-related DEGs and pathways. Finally, protein-protein interaction (PPI) network analysis was performed to identify the hub genes, and the DGIdb database was utilized to search for the candidate drugs for psoriasis.
Results
A total of 127 DEGs which were mostly associated with keratinization, keratinocyte differentiation, and epidermal cell differentiation biological processes were identified. Based on these GO terms, 3 modules (human skin, epidermis and cuticle differentiation, and enzyme activity) were constructed. Moreover, 9 risk subpathways such as steroid hormone biosynthesis, folate biosynthesis, and pyrimidine metabolism were screened. Finally, PPI network analysis demonstrated that CXCL10 was the hub gene with the highest degree, and CXCR2, CXCL10, IVL, OASL, and ISG15 were the potential gene targets of the drugs for treating psoriasis.
Conclusion
Psoriasis may be mostly caused by keratinization, keratinocyte differentiation, and epidermal cell differentiation; the pathogeneses were more related with pathways such as steroid hormone biosynthesis, folate biosynthesis, and pyrimidine metabolism. Besides, some psoriasis-related genes such as SPRR genes, HSD11B1, GGH, CXCR2, IVL, OASL, ISG15, and CXCL10 may be important targets in psoriatic therapy.
Similar content being viewed by others
Background
Psoriasis is a common, chronic, relapsing, and immune-mediated skin disease, prevalent in 2–4% of the total population [1]. Psoriasis is not purely a skin disorder and it may also affect many other organs and cause other diseases [2,3,4]. The cause of psoriasis in nearly one-third of the patients is hereditary, though it is often caused or influenced by environmental factors [5]. Oxidative stress, stress, and systemic corticosteroid withdrawal are thought to be the contributing factors to psoriasis [6]. Therefore, exploring the possible mechanisms of psoriasis is of great importance.
Previous studies have reported that psoriasis is generally caused by aberrant keratinocyte proliferation and differentiation, epidermal hyperplasia, angiogenesis, dendritic cells and neutrophils, infiltration of T lymphocytes, and elements of innate immunity [7]. Also, many studies focused on the identification of psoriasis susceptibility loci which were correlated with the pathogenesis. For instance, Zhang et al. investigated psoriasis susceptibility loci in psoriatic arthritis and a generalized pustular psoriasis cohort, and found 3 significant loci, and the most significant single nucleotide polymorphism (SNP) was rs7709212 in 5q33.3 [8]. Sheng et al. confirmed 4 known psoriasis susceptibility loci (IL12B, IFIH1, ERAP1 and RNF114; 2.30 × 10− 20 ≤ P ≤ 2.41 × 10− 7) and identified 3 new susceptibility loci: 4q24 (NFKB1) at rs1020760 (P = 2.19 × 10− 8), 12p13.3 (CD27-LAG3) at rs758739 (P = 4.08 × 10− 8) and 17q12 (IKZF3) at rs10852936 (P = 1.96 × 10− 8) [9]. Nevertheless, the molecular mechanisms of psoriasis remain to be fully understood.
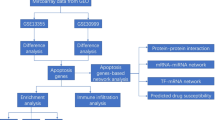
Nair et al. [10] identified psoriasis susceptibility loci and Swindell et al. [11] used genome-wide transcriptional analysis to compare gene expression patterns in human psoriatic lesions with those in 5 mouse models of psoriasis. However, they had not performed in-depth bioinformatics analyses to explore the rehabilitation mechanisms of psoriasis. Using the gene sets (GSE13355 and GSE14905) deposited by Nair et al. and Swindell et al., the gene expression profiling data related to psoriasis were used to generate the differentially expressed genes (DEGs) involved in psoriasis. Moreover, the gene ontology (GO) term enrichment analysis, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis, risk subpathway analysis, and construction of protein-protein interaction (PPI) network were all performed, and the DGIdb database was utilized to search for the candidate drugs for psoriasis. Finally, the DEGs and pathways mostly correlated with psoriasis were analyzed to investigate the pathogenesis. Therefore, our study provides a deep understanding of susceptibility genes in psoriasis and may provide appropriate drug targets for psoriatic therapies. A flow chart depicting the schematic of this study has been shown in Fig. 1.

Flow chart of this study
Results
Differentially expressed genes between normal and lesional groups
A total of 127 DEGs were screened between normal and lesional groups with |logFC| > 2 and adjusted P < 0.05, including 97 (76%) up-regulated DEGs and 30 (24%) down-regulated DEGs (Supplementary Table 1). The heatmap of the DEGs has been shown in Fig. 2, and the result shows that the two groups could be significantly separated, indicating that the results of the difference analysis were reliable.

Heatmap of DEGs. The red and green colors represent up-regulation and down-regulation, respectively
Principal component analysis of differentially expressed genes
As presented in Fig. 3, lesional and normal control samples were completely separated by the selected DEGs, meaning that the expression patterns of screened DEGs were specific and could be used to completely distinguish between lesional and normal control samples.

PCA between the psoriasis and normal control groups
Functional enrichment analysis of differentially expressed genes
Functional enrichment analyses of the GO terms and KEGG pathway were performed for both up-regulated and down-regulated DEGs. All the DEGs were mainly enriched in 48 GO terms, containing 30 ontology terms in biological process, and the top 10 GO terms in biological process have been shown in Table 1. Biological processes of DEGs were mostly associated with keratinization (GO: 0031424), keratinocyte differentiation (GO: 0030216), and epidermal cell differentiation (GO: 0009913). By integrating these GO terms using EnrichmentMap, 3 modules were obtained, which mostly associated with human skin (module 1), epidermis and cuticle differentiation (module 2), and enzyme activity (module 3) (Fig. 4). In the KEGG pathway analysis, the DEGs were enriched in 4 pathways and most significantly related to the chemokine signaling pathway (10 genes, hsa04062), cytokine-cytokine receptor interaction (9 genes, hsa04060), toll-like receptor signaling pathway (6 genes, hsa04620), and RIG-I-like receptor signaling pathway (4 genes, hsa04622) (Table 2). The enrichment analysis showed that small proline-rich protein (SPRR, including SPRR2C, SPRR1A and SPRR2B) genes were involved in all the biological processes related to epidermis and cuticle differentiation (Table 1). Moreover, CXC chemokine ligand (CXCL, including CXCL1, CXCL9 and CXCL10) genes were mostly involved in human skin-related GO terms and 4 KEGG pathways, and the C-X-C motif chemokine receptor 2 (CXCR2) was involved in the chemokine signaling pathway and cytokine-cytokine receptor interaction (Table 1 and Table 2).

Integration of GO terms for differentially expressed genes (DEGs). Red nodes represent GO terms. Green lines indicate that overlapping DEGs exist between two GO terms. Three circles represent three modules: module 1 mostly associates with skin, module 2 mostly associates with epidermis and cuticle differentiation, and module 3 mostly connects with enzyme activity
Risk subpathway enrichment analysis of differentially expressed genes
A total of 9 risk subpathways were identified by using the isubpathwayMiner package (Table 3). The three most significant subpathways were steroid hormone biosynthesis (path:00140_13) (P = 0.01561), folate biosynthesis (path:00790_4) (P = 0.01561), and pyrimidine metabolism (path:00240_21) (P = 0.019471). The enzyme 11beta-hydroxysteroid dehydrogenase type 1 (HSD11B1) is involved in steroid hormone biosynthesis, and the enzyme gamma-glutamyl hydrolase (GGH) is involved in folate biosynthesis.
Construction of protein-protein interaction network based on differentially expressed genes
To explore the interactions among psoriasis-related DEGs, the PPI network containing 71 nodes and 225 edges was constructed (Fig. 5). Of all the nodes, only 8 nodes were down-regulated DEGs, while the other 63 nodes were up-regulated DEGs. In this PPI network, the top 16 proteins with higher degrees were chosen, among which CXCL10 had the highest degree (Table 4).

The protein-protein interaction (PPI) network constructed on the interactions of protein molecules in psoriatic patients. Pink nodes represent the up-regulated DEGs; Green nodes represent the down-regulated DEGs; the edges represent the interactions between proteins
Drug-hub gene interaction
Using the 16 hub genes to explore the drug-gene interactions, 36 drugs for possibly treating psoriasis were compiled and selected (Fig. 6 and Table 5). Promising targets of these drugs included CXCR2, CXCL10, involucrin (IVL), 2'-5'-oligoadenylate synthetase like (OASL), and ISG15 ubiquitin-like modifier (ISG15).

Drug-hub gene interaction. Red nodes represent the up-regulated DEGs; Blue squares represent the drugs
Discussion
Psoriasis is a common skin disease, and it is an organ-specific autoimmune disease which is a common immune-mediated skin disease in adults [12]. Numerous studies have reported that psoriasis is generally caused by aberrant keratinocyte proliferation and epidermal hyperplasia [7, 13]. In this study, GO terms in biological processes (e.g. keratinization, keratinocyte differentiation, and epidermal cell differentiation) for DEGs were mostly associated with epidermis and cuticle differentiation, and the SPRR gene family were the most correlated DEGs. SPRR genes, consisting of two genes for SPRR1 (SPRR1A and SPRR1B), seven genes for SPRR2, and a single gene for SPRR3, encode a novel class of small proline-rich proteins that could be strongly induced during the differentiation of epidermal keratinocytes [14]. Kainu et al. found that three candidate gene clusters, the S100, SPRR, and PGLYRP genes, contained functionally interesting psoriasis candidate genes [15], which were consistent with our results. Besides, the KEGG pathway enrichment analysis showed that CXCR2 was involved in the chemokine signaling pathway and cytokine-cytokine receptor interaction. It has been shown that the expression of epidermal CXCR2 is increased in psoriasis, suggesting that activation of keratinocytes mediated by CXCR2 contributes to the characteristic epidermal changes observed in psoriasis [16]. The results of this study showed that CXCR2 was the potential gene target of the drugs for treating psoriasis. Moreover, proinflammatory cytokines are found to be associated with the progression of cerebral white matter injury (WMI) in preterm infants, and cytokine-receptor interaction may be critical in determining the effects of inflammation in the development of the disease, which further suggested that CXCR2 might have been related to the development of psoriasis via the chemokine signaling pathway and cytokine-cytokine receptor interaction.
Through risk subpathway enrichment analysis, subpathways such as steroid hormone biosynthesis, folate biosynthesis, and pyrimidine metabolism were identified, and HSD11B1 wass involved in steroid hormone biosynthesis, and GGH was involved in folate biosynthesis. HSD11B1 is the main regulator of tissue-specific effects of excessive circulating glucocorticoids, which may be related to inflammation [17, 18]. Thus, HSD11B1 was connected with psoriasis which has been always recognized as a chronic inflammatory disease of the skin. Additionally, Sevilla et al. showed that HSD11B1 was associated with osteoporosis, and HSD11B1/HSD11B2 were responsible for cortisol to cortisone interconversion [19]. Therefore, HSD11B1 might be recognized as the probable gene associated with psoriasis. Furthermore, Warren et al. reported that GGH was an important target gene of psoriasis which was involved in folate biosynthesis from our study [20]. Pyrimidine metabolism was another important subpathway identified in the current study. Zhou et al. constructed a ceRNA network to determine the regulatory roles of lncRNAs in psoriasis, and found that the upregulated mRNAs were mainly enriched in the pyrimidine metabolism, cell cycle, and cytokine-cytokine receptor interaction signaling pathways [21]. In summary, DEGs involved in risk subpathways were more likely related to psoriasis and might be important targets in psoriatic therapy. Also, all the subpathways identified by risk subpathway analysis were considered as the most relevant pathways with psoriasis.
The PPI network analysis showed that protein with the highest degree was encoded by CXCL10, a member of the CXC chemokine family of cytokines [22]. The results of this study showed that CXCL10 was the potential gene target of the drugs for treating psoriasis. CXCL10 has been reported to be expressed in many Th1-type human inflammatory diseases, including skin diseases (e.g. psoriasis) [23]. A recent study suggested that the decrease in serum CXCL10 level over time was related to the new onset of psoriatic arthritis in patients with psoriasis [24], and our study revealed a same result that CXCL10 was up-regulated in psoriasis. In the present study, the CXC chemokine family of cytokines (containing CXCL1, CXCL9, and CXCL10) was mostly enriched in human skin-related GO terms and KEGG pathways such as the chemokine signaling pathway and cytokine-cytokine receptor interaction, implying that the gene family might be important in regulating psoriasis. Luster et al. reviewed that chemokines-chemotactic cytokines could mediate inflammation, which might be the reason for the association between CXC chemokine family and psoriasis [25]. IVL, OASL, and ISG15 were also the potential gene targets of the drugs for treating psoriasis in this study. Chen et al. found that IVL was related to the early-onset plaque psoriasis [26]. The expression pattern of OASL may offer useful information for treating autoimmune diseases and chronic infections [27]. In addition, Raposo et al. observed significant overexpression of 16 antiviral genes in lesional psoriatic skin, with more than two-fold increase in ISG15 [28].
However, there are some limitations in this study. For instance, the microarray data was obtained from the GEO database, and not generated by the authors. Therefore, further experiments should be conducted to verify whether these target genes can be used in the clinical treatment of psoriasis.
Conclusions
In the current study, risk subpathway analysis based on DEGs was considered a useful method to identify pathways mostly related to psoriasis. Psoriasis may be mostly caused by keratinization, keratinocyte differentiation, and epidermal cell differentiation and the pathogeneses were more related to pathways such as steroid hormone biosynthesis, folate biosynthesis, and pyrimidine metabolism. Besides, some psoriasis-related genes such as SPRR genes, HSD11B1, GGH, CXCR2, IVL, OASL, ISG15, and CXCL10 may be important targets in psoriatic therapy. Therefore, our study explored the pathogeneses of psoriasis in depth and provided probable target genes for psoriatic therapy.
Methods
Microarray data
Gene expression data of human mRNA about psoriasis research (GSE13355 and GSE14905) were obtained from the Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) database. There were 180 samples in GSE13355, including 64 healthy control samples without psoriasis and 116 experimental samples with psoriasis. We chose 64 healthy control samples without psoriasis and 58 skin samples from psoriasis patients in this study. Also, 82 samples from GSE14905, including 21 healthy control samples without psoriasis and 61 experimental samples with psoriasis, among which 21 healthy control samples without psoriasis and 33 skin samples from psoriasis patients were selected in this study. All samples were detected through the Affymetrix Human Genome U133 Plus 2.0 array platform. The sample basic statistical information of GSE13355 and GSE14905 datasets have been shown in Supplementary Table 2 and Supplementary Table 3.
Data preprocessing and screening of differentially expressed genes
In order to obtain the common gene expression matrix and eliminate the heterogeneity among datasets, we used the ComBat function of sva package [29] of R language to eliminate batch effect between datasets. In addition, the Affy package [30] was applied for preprocessing the obtained data by conducting normalization, background correction and expression calculation. Then the probes were annotated by matrix data combined with chip platform annotation file. The average value of diverse probes would be considered as the eventual expression level of gene if they corresponded to the identical mRNA. Finally, a total of 54,675 probes were mapped to 19,851 gene symbols without repetition.
After data preprocessing, the limma package [31] of R language was utilized to screen DEGs. The false discovery rate (FDR) of Benjamini and Hochberg (BH) method was applied to adjust p-values for multiple comparisons. The significant differentially expressed cut-off was set as |logFC| > 2 and adjusted P < 0.05. Finally, a heatmap was drawn to observe the clustering of samples.
Principal component analysis of differentially expressed genes
Principal component analysis (PCA), a multivariate regression analysis, was used to distinguish samples with multiple measurements [32]. A PCA of DEGs was conducted using the ggord package (version: 1.1.4) in R language in the present study. The PCA graph was then obtained, in which DEGs were considered as variables and the differences between normal control and lesional samples were observed.
GO and KEGG enrichment analysis
The enrichment analysis of DEGs was carried out by utilizing the online software DAVID [33], in which species was set as human and the other parameters were the default values. GO terms with a cut-off of P < 0.05 and gene count ≥5 were chosen. Likewise, the KEGG pathways with P < 0.05 were selected. Then a plug-in EnrichmentMap in Cytoscape [34] was used to integrate the GO terms with the cutoff of P < 0.05 and overlap coefficient larger than 0.5.
Risk subpathway screening for differentially expressed genes
To obtain more important genes and pathways in psoriasis, the iSubpathwayMiner package (version: 3.0) [35] in R language (version: 3.4.3) was selected to identify the risk subpathways for DEGs. Moreover, the subpathways with P < 0.05 were screened, which reflected the pathways involved in psoriasis more clearly.
Protein-protein interaction network construction
To further analyze the impacts of DEGs on psoriasis, the PPI network was constructed among various DEGs. Also, the online software STRING was used to analyze the interactions of proteins encoded by DEGs [36], and the PPI score was set as 0.4 (referred as median confidence). Then the Cytoscape software [34] (https://cytoscape.org/) was utilized to visualize the PPI network. Finally, the degree of each DEG was obtained by analyzing the topological structure of the PPI network.
Drug-hub gene interaction
The hub genes with high degree (degree ≥10) of connectivity in PPI were selected as the promising targets for searching drugs through the DGIdb database (version: 3.0.3, http://www.dgidb.org/). This database contains drug-gene interaction data from 30 disparate sources including ChEMBL, DrugBank, Ensembl, NCBI Entrez, PharmGKB, PubChem, clinical trial databases, and literature in NCBI PubMed. The results of this process were arranged such that each entry was a specific drug-gene interaction associated with its source link [37]. The identified target network was visualized using the Cytoscape software.
Availability of data and materials
All data generated or analysed during this study are included in this published article.
Abbreviations
- GEO:
-
Gene Expression Omnibus
- DEGs:
-
Differentially expressed genes
- GO:
-
Gene ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- PPI:
-
Protein-protein interaction
- HSD11B1 :
-
11beta-hydroxysteroid dehydrogenase type 1
- GGH :
-
Gamma-glutamyl hydrolase
References
Mease P, Palmer J, Hur P, Strober B, Lebwohl M, Karki C, et al. Utilization of the validated psoriasis epidemiology screening tool to identify signs and symptoms of psoriatic arthritis among those with psoriasis: a cross-sectional analysis from the US-based Corrona psoriasis registry. J Eur Acad Dermatol Venereol. 2019;33(5):886–92.
De CS, Caldarola G, Moretta G, Piscitelli L, Ricceri F, Prignano F. Moderate-to-severe psoriasis and pregnancy: impact on fertility, pregnancy outcome and treatment perspectives. Giornale italiano di dermatologia e venereologia: organo ufficiale, Societa italiana di dermatologia e sifilografia. 2019;154(3):305–14.
Takeshita J, Grewal S, Langan SM, Mehta NN, Ogdie A, Van Voorhees AS, et al. Psoriasis and comorbid diseases: epidemiology. J Am Acad Dermatol. 2017;76(3):377–90.
Michalek I, Loring B, John S. A systematic review of worldwide epidemiology of psoriasis. J Eur Acad Dermatol Venereol. 2017;31(2):205–12.
Kunz M, Simon JC, Saalbach A. Psoriasis: obesity and fatty acids. Front Immunol. 2019;10:1807.
Yang H, Tan Q, Chen G, Chen J, Fu Z, Ren F, et al. Plasma retinol as a predictive biomarker of disease activity and response to acitretin monotherapy in children with generalized pustular psoriasis. J Eur Acad Dermatol Venereol. 2020;34(6):e270–2.
Lowes MA, Bowcock AM, Krueger JG. Pathogenesis and therapy of psoriasis. Nature. 2007;445(7130):866–73.
Zhang Z, Xu JH. Investigation of psoriasis susceptibility loci in psoriatic arthritis and a generalized Pustular psoriasis cohort. J Investigative Dermatol Symposium Proc. 2018;19(2):S83–s5.
Sheng Y, Jin X, Xu J, Gao J, Du X, Duan D, et al. Sequencing-based approach identified three new susceptibility loci for psoriasis. Nat Commun. 2014;5:4331.
Nair RP, Duffin KC, Helms C, Ding J, Stuart PE, Goldgar D, et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-κB pathways. Nat Genet. 2009;41(2):199.
Swindell WR, Johnston A, Carbajal S, Han G, Wohn C, Lu J, et al. Genome-wide expression profiling of five mouse models identifies similarities and differences with human psoriasis. PLoS One. 2011;6(4):e18266.
El-Reshaid K, Shaima A-B. Long-term maintenance therapy with cyclosporine a in adults with generalized pustular psoriasis. J Drug Delivery Therapeutics. 2019;9(5):19–21.
Zhang X, Yin M, Zhang L-j. Keratin 6, 16 and 17—Critical Barrier Alarmin Molecules in Skin Wounds and Psoriasis. Cells. 2019;8(8):807.
Gibbs S, Fijneman R, Wiegant J, van Kessel AG, van de Putte P, Backendorf C. Molecular characterization and evolution of the SPRR family of keratinocyte differentiation markers encoding small proline-rich proteins. Genomics. 1993;16(3):630–7.
Kainu K, Kivinen K, Zucchelli M, Suomela S, Kere J, Inerot A, et al. Association of psoriasis to PGLYRP and SPRR genes at PSORS4 locus on 1q shows heterogeneity between Finnish. Swedish Irish Families  Experimental Dermatology. 2008;18(2):109–15.
Kondo S, Yoneta A, Yazawa H, Kamada A, Jimbow K. Downregulation of CXCR-2 but not CXCR-1 expression by human keratinocytes by UVB. J Cell Physiol. 2000;182(3):366–70.
D’Attilio L, Diaz A, Santucci N, Bongiovanni B, Gardenez W, Marchesini M, et al. Levels of inflammatory cytokines, adrenal steroids, and mRNA for GRalpha, GRbeta and 11betaHSD1 in TB pleurisy. Tuberculosis (Edinb). 2013;93(6):635–41.
Morgan SA, McCabe EL, Gathercole LL, Hassan-Smith ZK, Larner DP, Bujalska IJ, et al. 11β-HSD1 is the major regulator of the tissue-specific effects of circulating glucocorticoid excess. Proc Natl Acad Sci. 2014;111(24):E2482–91.
Sevilla LM, Pérez P. Glucocorticoids and glucocorticoid-induced-Leucine-zipper (GILZ) in psoriasis. Front Immunol. 2019;10:2220.
Warren RB, Smith RL, Campalani E, Eyre S, Smith CH, Barker JN, et al. Outcomes of methotrexate therapy for psoriasis and relationship to genetic polymorphisms. Br J Dermatol. 2009;160(2):438–41.
Zhou Q, Yu Q, Gong Y, Liu Z, Xu H, Wang Y, et al. Construction of a lncRNA-miRNA-mRNA network to determine the regulatory roles of lncRNAs in psoriasis. Exp Therapeutic Med. 2019;18(5):4011–21.
Strieter R, Kunkel S, Arenberg D, Burdick M, Polverini P. Interferon [gamma]-inducible protein-10 (IP-10), a member of the CXC chemokine family, is an inhibitor of angiogenesis. Biochem Biophys Res Commun. 1995;210(1):51–7.
Dufour JH, Dziejman M, Liu MT, Leung JH, Lane TE, Luster AD. IFN-γ-inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10 in effector T cell generation and trafficking. J Immunol. 2002;168(7):3195–204.
Abji F, Lee K, Math M, Pollock R, Machhar R, Cook R, et al. Declining levels of serum CXCL10 over time are associated with new onset of psoriatic arthritis in patients with psoriasis: a new biomarker? Br J Dermatol. 2020. https://doi.org/10.1111/bjd.18940. [Published online ahead of print, 2020 Feb 9].
Epstein FH, Luster AD. Chemokines—chemotactic cytokines that mediate inflammation. N Engl J Med. 1998;338(7):436–45.
Chen H, Toh TK, Szeverenyi I, Ong RT, Theng CT, McLean WH, et al. Association of skin barrier genes within the PSORS4 locus is enriched in Singaporean Chinese with early-onset psoriasis. J Investigative Dermatol. 2009;129(3):606–14.
Choi UY, Kang JS, Hwang YS, Kim YJ. Oligoadenylate synthase-like (OASL) proteins: dual functions and associations with diseases. Exp Mol Med. 2015;47:e144.
Raposo RA, Gupta R, Abdel-Mohsen M, Dimon M, Debbaneh M, Jiang W, et al. Antiviral gene expression in psoriasis. J Eur Acad Dermatol Venereol. 2015;29(10):1951–7.
Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28(6):882–3.
Gautier L, Cope L, Bolstad BM, Irizarry RA. Affy--analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20(3):307–15.
Smyth GK, Michaud J, Scott HS. Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics. 2005;21(9):2067–75.
Zhao W, Wang D, Zhao J, Zhao W. Bioinformatic analysis of retinal gene function and expression in diabetic rats. Exp Therapeutic Med. 2017;14(3):2485–92.
Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, et al. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4(5):P3.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Li C, Li MC. Package ‘iSubpathwayMiner’; 2013.
Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, et al. STRING v9. 1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41(D1):D808–D15.
Kirk J, Shah N, Noll B, Stevens CB, Lawler M, Mougeot FB, et al. Text mining-based in silico drug discovery in oral mucositis caused by high-dose cancer therapy. Supportive care in cancer : official journal of the Multinational Association of Supportive Care in Cancer. 2018;26(8):2695–705.
Acknowledgements
None.
Funding
This work was supported by Program of Hunan Provincial Health Commission [grant number 20200341]; and Key Laboratory Fund of Hunan Province [grant number 2018tP1028].
Author information
Authors and Affiliations
Contributions
Conception and design of the research: YQL; acquisition of data: YYL, JC; analysis and interpretation of data: YYL, JC; statistical analysis: ZX; drafting the manuscript: YQL; revision of manuscript for important intellectual content: BZ. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Differentially expressed genes between normal and lesional groups.
Additional file 2: Table S2.
The sample basic statistic information of GSE13355 dataset.
Additional file 3: Table S3.
The sample basic statistic information of GSE14905 dataset.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Luo, Y., Luo, Y., Chang, J. et al. Identification of candidate biomarkers and pathways associated with psoriasis using bioinformatics analysis. Hereditas 157, 30 (2020). https://doi.org/10.1186/s41065-020-00141-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41065-020-00141-1