
Abstract
Before ‘cancer interception’ was first advocated, ‘interceptor molecules’ had been conceived as a sub-category of preventive agents that interfered with the earliest initiation steps in carcinogenesis. Three decades ago, a seminal review cataloged over fifty synthetic agents and natural products that were known or putative interceptor molecules. Chlorophylls and their derivatives garnered much interest based on the potent antimutagenic activity in the Salmonella assay, and the subsequent mechanistic work that provided proof-of-concept for direct molecular complexes with planar aromatic carcinogens. As the ‘interceptor molecule’ hypothesis evolved, mechanistic experiments and preclinical studies supported the view that chlorophylls can interact with environmental heterocyclic amines, aflatoxins, and polycyclic aromatic hydrocarbons to limit their uptake and bioavailability in vivo. Support also came from human translational studies involving ultralow dose detection in healthy volunteers, as well as intervention in at-risk subjects. Antimutagenic and antigenotoxic effects of natural and synthetic chlorophylls against small alkylating agents also highlighted the fact that non-interceptor mechanisms existed. This gave impetus to investigations broadly related to free radical scavenging, anti-inflammatory effects, immune modulation and photodynamic therapy. Therapeutic aspects of chlorophylls also were investigated, with evidence for cell cycle arrest and apoptosis in human cancer cells. As the science has evolved, new mechanistic leads continue to support the use and development of chlorophylls and their porphyrin derivatives for cancer interception, beyond the initial interest as interceptor molecules.
Similar content being viewed by others

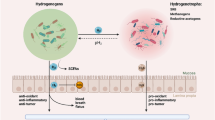

Background
Long before the term 'cancer interception' came into vogue [1, 2], 'interceptor molecules' already had been conceptualized [3], incorporating 'desmutagens' and inhibitors that might prevent the formation of carcinogens [4,5,6,7,8]. Harman and Shankel [3] noted that antimutagens could act at multiple levels, including the following: (i) prevention of mutagen formation; (ii) interception of mutagens via cellular or tissue organization; (iii) interception of mutagens by metabolites or enzymes present in cells; (iv) neutralization or removal of pre-mutagenic lesions in DNA by chemical compounds; and (v) activation of mechanisms that enhanced error-free DNA repair, blocked error-prone DNA repair, or augmented the metabolic inactivation of mutagens. The diverse range of compounds surveyed included N-acetyl-L-cysteine, acylglucosylsterols, albumins, allyl sulfides, p-aminobenzoic acid, aromatic isothiocyanates, ascorbic acid, bilirubin and biliverdin, bioflavonoids, butylated hydroxyanisole and butylated hydroxytoluene, caffeic acid, calcium, L-carnosine, β-carotene, catechins, chalcones, chlorogenic acids, creatine and creatinine, curcumin, diallyl sulfides, α- and β-dicarbonyls, dithiolthiones, ellagic acid, eugenol, fatty acids, ferulic acid, fiber, gallic acid, γ-glutamylcysteine, glutathione, L-histidine, hydroxychavicol, hypotaurine, imidazole-4-acetate, mucins, myricetin, ovothiols, pantetheine, polyamines, polyphenols, polyunsaturated fatty acids, quercetin, retinoids, tannins, taurine, thiols, tocopherols, uric acid, vitamins, and miscellaneous other agents [3]. The latter review also included chlorophylls as putative interceptor molecules [3], as these phytochemicals will be discussed here in greater detail, given that the field has continued to evolve over the intervening three decades.
Review
Chlorophylls as interceptor molecules
Hayatsu and colleagues first reported on the antimutagenic activities of chlorophylls and other porphyrins in the Salmonella mutagenicity assay [9,10,11], which subsequently was confirmed by others [12,13,14,15]. A seminal report [10] described molecular complex formation in vitro between a heterocyclic amine mutagen and chlorophyllin (CHL), the water-soluble derivative of natural chlorophyll a (Chla). Although evidence was lacking in a preclinical cancer model, important groundwork had been laid for the ‘interceptor molecule’ hypothesis, with the possibility that molecular complexes might lower carcinogen uptake and systemic bioavailability after oral exposure. The appeal of such a mechanism immediately was apparent, given the ubiquitous presence of spinach and other chlorophyll-rich green leafy vegetables in the human diet [16,17,18]. Moreover, the mechanism implied broad applicability against a wide range of planar aromatic carcinogens that were known from the literature, including cooked meat heterocyclic amines, environmental polycyclic aromatic hydrocarbons, and dietary aflatoxins [9,10,11,12,13,14,15]. These aspects will be reviewed in the following sections.
Interception of heterocyclic amines by chlorophylls
Spectrophotometric titration studies provided evidence for molecular complex formation between CHL and 2-amino-3-methylimidazo [4,5-f] quinoline (IQ), as well as with a dozen other dietary heterocyclic amine mutagens [19,20,21,22]. An isosbestic point indicated the presence of a 1:1 complex in some cases, which was corroborated via the mole ratio plot, whereas other interactions were more consistent with 2:1 stoichiometry. Dissociation/binding constants and docking scores in silico supported reversible complexes involving multiple π-π (stacking) interactions stabilized by van der Waals and electrostatic bonds, which inhibited IQ-DNA binding in vitro [19]. An inverse correlation was observed between the binding constant (Kb) of the complex and the antimutagenic potency in the Salmonella assay, i.e., the more stable the interaction, the lower the concentration of CHL needed to inhibit mutagenicity [20]. Notably, these findings were extended to natural chlorophylls, chlorins, tetrapyrroles, and other porphyrins [21].
Based on the hypothesis that molecular complexes would limit carcinogen uptake and bioavailability, corroborative experiments were conducted in vivo [23,24,25]. Co-administration of CHL and 14C-labeled IQ to the rat via single oral gavage inhibited IQ-DNA adduct formation significantly in the liver and colon [23, 24], and IQ-derived radiolabel was reduced in liver, bile and urine while being increased simultaneously in the feces. Co-injection of CHL with 14C-labeled IQ into isolated loops of intestine in situ also reduced the absorption of IQ from the gut, compared with IQ alone [23]. Pretreatment with CHL in the drinking water for 2 days before administering IQ by single oral gavage increased the excretion of conjugated (detoxified) mutagens in the urine and feces [25]. Finally, administration of IQ three times per week by oral gavage, while providing CHL in the drinking water, inhibited significantly the total tumor burden at 52 weeks in the male F344 rat [26].
This was the first evidence in support of the anticarcinogenic activity of CHL in vivo, together with the work by Hasegawa et al. on the inhibition of mammary carcinogenesis in female rats treated with the heterocyclic amine 2-amino-1-methyl-6-phenylimidazo [4,5-b] pyridine (PhIP) [27]. Findings from the author’s laboratory are presented to highlight key aspects of the discussion above (Fig. 1).

Chlorophyllin (CHL), a water-soluble derivative of natural chlorophyll, is an interceptor molecule of heterocyclic amines. a Spectrophotometric titration studies identified molecular complex formation between CHL and the cooked meat mutagen 2-amino-3-methylimidazo [4,5-f] quinoline (IQ). b Benesi-Hildebrand plot and molecular docking in silico of the CHL-IQ complex. Dissociation constants also were defined for other dietary heterocyclic amines, with complexes involving 1:1 or 2:1 stoichiometry. c In vitro, the kinetics of IQ-DNA binding indicated competitive inhibition by CHL. d In vivo, co-administration of CHL and IQ by single oral gavage reduced IQ-DNA adducts significantly in the rat liver (*P < 0.05). e Inhibition of IQ-induced tumorigenesis in the rat following co-treatment with CHL and IQ for 1 year. From the author’s published reports [19,20,21,22,23,24,25,26]
Interception of aflatoxins by chlorophylls
In addition to its potent antimutagenic activity towards aflatoxin B1 (AFB1), CHL also was highly effective against the direct-acting intermediate AFB1–8,9-epoxide in the absence of a metabolic activation system [13, 14]. Molecular docking in silico and spectrophotometric titration experiments supported a dissociation constant of Kd = 1.4 ± 0.4 μM for the AFB1-CHL complex. In rainbow trout, AFB1-CHL coadministration in the diet produced concentration-dependent inhibition of AFB1-DNA adducts in the liver at 2 weeks by CHL, and predicted precisely the reduced incidence of hepatocellular carcinoma 9 months later [28,29,30,31].
Notably, hepatic AFB1-DNA adducts were reduced significantly in the rat and rainbow trout following simultaneous oral gavage administration of the carcinogen with either CHL or natural Chla [31, 32]. The inhibition of multiorgan carcinogenesis occurred independently of changes in hepatic enzyme activities, supporting a mechanism involving complex-mediated reduction of carcinogen uptake in vivo [31, 32].
In a landmark report [33], the bioavailability of aflatoxin was reduced significantly in human volunteers by CHL or natural Chla isolated from spinach, using the sensitivity of accelerator mass spectrometry to detect microdosing levels in plasma and urine [34]. These findings extended prior work demonstrating that CHL intervention via an oral supplement reduced aflatoxin-DNA adducts in individuals at high risk for liver cancer [35, 36]. Highlights from the associated literature illustrate the continuum from mechanistic studies in vitro, to preclinical validation in animal models, and subsequent human translation (Fig. 2; silhouettes indicate trout, rat, and human translational aspects).

CHL and natural chlorophyll a (Chla) act as interceptor molecules against aflatoxin B1 (AFB1). a Antimutagenic activity of CHL against AFB1–8,9-epoxide in the Salmonella assay, in the absence of a metabolic activation system, with complex formation supported by molecular docking in silico. b CHL inhibits AFB1-DNA binding and hepatocellular carcinoma in rainbow trout, and is more effective than Chla at inhibiting AFB1-DNA adducts in trout liver (****P < 0.0001, ***P < 0.001). c CHL and Chla inhibit AFB1-DNA adducts in rat liver (**P < 0.01). d Accelerator mass spectrometry revealed that oral administration of CHL or Chla both lowered AFB1 uptake and bioavailability in human volunteers. This work highlights the continuum from mechanistic to preclinical to human translational findings [14, 28,29,30,31,32,33,34,35,36]
Interception of polycyclic aromatic hydrocarbons by chlorophylls
Based on prior work [37] establishing the inhibitory actions of CHL and related pyrrole pigments against the mutagenicity of benzo [a] pyrene (BaP), detailed molecular mechanisms were investigated in vitro [15]. In the Salmonella assay, CHL inhibited the mutagenic activity of BaP in the presence of a metabolic activation system, and it was especially effective against the direct-acting ultimate carcinogen benzo [a]pyrene-7,8-dihydrodiol-9,10-epoxide (BPDE) in the absence of exogenous mammalian liver enzymes. Time-dependent hydrolysis of BPDE to inactive tetrols was observed in the presence of 5 μM CHL, whereas molecular complex formation with the procarcinogen (BaP) and the inhibition of cytochrome P450-related enzymatic activities necessitated higher CHL concentrations (> 100 μM). At these concentrations, CHL inhibited NADPH-cytochrome P450 reductase activity, rather than binding directly to the active site of cytochrome P450. Molecular models of the BPDE:CHL complex revealed minimization energies in the range − 16.9 to − 20.8 kcal/mol, with multiple π-π interactions of the overlapping aromatic ring systems, and the epoxide moiety of BPDE oriented towards acid (carboxyl) groups or the methylene bridge in CHL. It was concluded that the primary mechanism of CHL towards BaP in vivo involved preferential molecular complex formation with BPDE, leading to the rapid degradation of the ultimate carcinogen [15].
In female ICR mice, oral gavage dosing of CHL 30 min before either BaP or BPDE were given by topical administration resulted in significant inhibition of skin tumorigenesis, and CHL was rapidly distributed to the skin and other tissues [38]. The authors concluded that, under the experimental conditions used, the inhibition of skin carcinogenesis in mice was consistent with the interceptor molecule hypothesis.
Dibenzo [def,p] chrysene (DBC) – formerly known as dibenzo [a,l] pyrene – is a structurally-related environmental agent of BaP that has greater carcinogenic potency due to the presence of a ‘fjord’ region rather than a ‘bay’ region, which stabilizes the reactive epoxide intermediate [39]. In the rainbow trout, hepatic DBC-DNA adducts were inhibited significantly when the carcinogen was co-administered in the diet for 2 weeks with either CHL or natural Chla [40]. Spectrophotometric titration studies supported a direct interaction between DBC and either CHL or Chla. Thus, a 2:1 complex was observed for CHL-DBC, with Kd1 = 1.38 μM and Kd2 = 1.17 μM, whereas the 2:1 Chla-DBC complex had Kd1 = 4.44 μM and Kd2 = 3.30 μM [41]. Pharmacokinetic data revealed that CHL and Chla both lowered the systemic bioavailability of DBC to the liver and other tissues, consistent with the effective antitumor activity in this animal model [41, 42]. In the mouse, CHL provided effective chemoprotection in a DBC-induced transplacental carcinogenesis model, and supported a mechanism involving complex-mediated reduction of carcinogen uptake [43]. These findings suggest that additional studies are warranted on the beneficial effects of CHL and Chla towards other environmental carcinogens and complex mixtures [44]. Mechanistic aspects for CHL and Chla towards polycyclic aromatic hydrocarbons in vitro and in vivo were highlighted in Fig. 3.

Chlorophylls as interceptor molecules of environmental polycyclic aromatic hydrocarbons. a Interaction of CHL with benzo [a]pyrene-7,8-dihydrodiol-9,10-epoxide (BPDE). Absorption spectra for BPDE (dashed line), CHL (dotted line), and BPDE+CHL (solid line) in Tris-HCl buffer, pH 7.4, 20 °C. Inset: difference spectrum for the BPDE/CHL complex. b Time-dependent hydrolysis of BPDE in presence and absence of CHL. Upper panel: the hydrolysis of 10 μM BPDE to tetrols was recorded at 5-min intervals in Tris-HCl buffer, pH 7.4, 20 °C. Lower panel: repeat of the above experiment in the presence of 5 μM CHL. Numbers adjacent to spectra indicate the time in minutes at which each spectrum was recorded. The pseudo-first-order hydrolysis rate constant of BPDE to tetrols was 1.76 ± 0.67 in buffer and 8.37 ± 0.82 in buffer containing CHL (kH (s− 1) × 103). c Energy minimized molecular model of the BPDE:CHL complex. Initial structures of CHL (top) and BPDE (bottom) were constructed using MM2 force field parameters and conjugate gradient methods. Complexes were obtained by first energy minimizing the structures of CHL and BPDE separately, then placing each molecule within Van der Waals radii and energy minimizing the corresponding complex. After multiple iterative docking experiments, complexes consistently had the epoxide oriented towards acid (carboxyl) groups or the methylene bridge in CHL, with minimization energies in the range − 16.9 to − 20.8 kcal/mol. Complexes were minimized to a gradient of 0.001 kcal/mol and calculations were performed using HyperChem (Release 2, Autodesk). d Natural Chla complexes with dibenzo [def,p] chrysene (DBC). e Chla and CHL lower the bioavailability of DBC and (inset) DBC-DNA adducts detected in trout liver (***P < 0.001). f Concentration-dependent inhibition of DBC-induced tumorigenesis in the rainbow trout. From published reports [15, 40,41,42]
Non-interceptor mechanisms of chlorophylls
Despite the simplicity and attractive nature of the interceptor molecular hypothesis, it was known from early antimutagenicity work in vitro that CHL also was effective against N-methyl-N′-nitro-N-nitrosoguanidine [44]. This small alkylating agent lacks the aromatic ring system for stable molecular complexes involving multiple π-π interactions. Investigations with other small non-aromatic compounds, such as 1,2-dimethylhydrazine, azoxymethane and diethylnitrosamine, identified tumor modulatory activity even in post-initiation protocols, in which the carcinogen treatment phase had been completed before the addition of CHL or Chla [45,46,47,48,49,50,51,52,53,54,55]. In some reports, the apparent tumor promoting activities may have been related to impurities in commercial CHL preparations [56, 57], which contained a mixture of sodium-potassium salts and chlorins [28], sometimes detectable in human plasma during clinical trials [35].
However, further investigation confirmed that CHL indeed exhibited mechanisms beyond molecular complex formation. The large tetrapyrrole macrocycle found in chlorophylls, chlorins, and other porphyrins has a high degree of unsaturation, which implicated free radical scavenging, antioxidant, anti-inflammatory, immune-modulatory and photodynamic properties. These aspects have been discussed previously for chlorophyllin and related porphyrins, including the effects on Wnt/β-catenin signaling and other deregulated pathways in cancer [58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76].
To examine mechanisms that go beyond molecular complex formation, human colon cancer cells were incubated with CHL across a broad range of concentrations [77, 78]. Fluorescence-activated cell sorting (FACS) analysis revealed that CHL-treated cells underwent S-phase arrest, and at higher concentrations a sub-G1 peak was detected, indicative of apoptosis [77]. Cells entering S-phase arrest exhibited a concentration-dependent loss of bromodeoxyuridine (BrdU) incorporation in FACS-based pulse-chase experiments. This was analogous to prior studies with chemotherapeutic drugs, such as hydroxyurea, that inhibited ribonucleotide reductase (RNR) activity by scavenging the tyrosyl radical involved in the catalytic mechanism [79,80,81,82,83]. Subsequently, CHL was confirmed to downregulate the expression of the large and small subunits of RNR, namely R1, R2 and p53R2, and to directly inhibit RNR enzymatic activity [78].
At higher concentrations, colon cancer cells exhibited the hallmarks of apoptosis, such as membrane blebbing and nuclear condensation, without the classical apoptotic ‘ladder’ in gel-based experiments [77]. Although a change in mitochondrial membrane potential was detected (∆ψm), this was not accompanied by the release of cytochrome c or the activation of Caspase-9, Caspase-3, and poly (ADP-ribose) polymerase (PARP) cleavage. However, a cytochrome c-independent apoptotic mechanism was confirmed that involved Caspase-8, Caspase-6, the release from mitochondria of apoptosis inducing factor (AIF), and the cleavage of nuclear lamins [77]. This work advanced key concepts beyond prevention by an ‘interceptor molecule’ towards late-stage cancer interception, via mechanisms that impacted cell cycle regulation and apoptosis (Fig. 4).

CHL triggers cell cycle arrest and apoptosis in human colon cancer cells. a In CHL-treated HCT116 cells, fluorescence-activated cell sorting (FACS) identified cells arrested in S-phase, with the appearance of a sub-G1 peak at higher CHL concentrations, indicative of apoptosis. b S-phase arrest by CHL was concentration dependent. c FACS analysis combined with bromodeoxyuridine (BrdU) pulse-chase experiments corroborated the S-phase arrest, with BrdU incorporation localized to G0/G1 and G2/M lower quadrants. d The floating cell number increased with CHL concentration, and cells exhibited hallmarks of apoptosis such as membrane blebbing and nuclear condensation, coinciding with reduced mitochondrial membrane potential (∆ψm), arrow. e Detailed mechanistic studies excluded the pathway involving cytochrome c, Caspase-9, Caspase-3 and poly (ADP-ribose) polymerase (PARP) cleavage, and implicated Capase-8, Caspase-6, cleavage of nuclear lamins, and apoptosis inducing factor (AIF) released from mitochondria. At lower CHL concentrations, S-phase arrest involved the inhibition of ribonucleotide reductase, possibly by scavenging the tyrosyl radical in the enzyme active site, akin to the anticancer drug hydroxyurea. Synopsis of author’s prior mechanistic work [77, 78]
Finally, epigenetic avenues remain to be pursued in vivo, beyond the downregulation of histone deacetylases in hamster-cheek pouch carcinomas by CHL [84], and the altered microRNA signatures in PhIP-induced rat colon tumors after feeding chlorophyll-rich spinach [85]. Antiviral aspects of CHL also are highly noteworthy [86].
Conclusions
In conclusion, chlorophylls can act as interceptor molecules of environmental carcinogens and mutagens, but also exhibit additional mechanisms that impact the continuum of cancer initiation, promotion and progression. As the science evolves, interesting new mechanistic leads point to the potential use of chlorophylls and their derivatives for cancer interception, in its broadest context [1]. Thus, further research appears to be warranted, beyond the historical clinical applications and the current use of such natural and synthetic agents as food additives [87,88,89,90], directed towards environmental carcinogens and the human translational aspects [91,92,93,94,95,96,97,98,99,100].
Availability of data and materials
Not applicable – data are available in the public domain.
Abbreviations
- AFB1 :
-
Aflatoxin B1
- AIF:
-
Apoptosis inducing factor
- BaP:
-
Benzo [a]pyrene
- BPDE:
-
Benzo [a]pyrene-7,8-dihydrodiol-9,10-epoxide
- BrdU:
-
Bromodeoxyuridine
- CHL:
-
Chlorophyllin
- Chla :
-
Chlorophyll a
- DBC:
-
Dibenzo [def,p]chrysene
- FACS:
-
Fluorescence-activated cell sorting
- IQ:
-
2-amino-3-methylimidazo [4,5-f]quinoline
- Kb:
-
Binding constant
- Kd:
-
Dissociation constant
- PARP:
-
Poly (ADP-ribose)polymerase
- PhIP:
-
2-amino-1-methyl-6-phenylimidazo [4,5-b]pyridine
- RNR:
-
Ribonucleotide reductase
References
Blackburn EH. Cancer interception. Cancer Prev Res (Phila). 2011;4:787–92.
Lippmann SM, Abate-Shen C, Maresso KLC, Colditz GA, Dannenberg AJ, Davidson NE, Disis ML, DuBois RN, Szabo E, Giuliano AR, Hait WN, Lee JJ, Kensler TW, Kramer BS, Limburg P, Maitra A, Martinez ME, Rebbeck TR, Schmitz KH, Vilar E, Hawk ET. AACR white paper: shaping the future of cancer prevention – a roadmap for advancing science and public health. Cancer Prev Res. 2018;11:735–78.
Hartman PE, Shankel DM. Antimutagens and anticarcinogens: a survey of putative interceptor molecules. Environ Mol Mutagen. 1990;15:145–82.
Osawa T, Ishibashi H, Namiki M, Kada T. Desmutagenic actions of ascorbic acid and cysteine on a new pyrrole mutagen formed by the reaction between food additives; sorbic acid and sodium nitrite. Biochem Biophys Res Commun. 1980;95:835–41.
Morita K, Kada T, Namiki M. A desmutagenic factor isolated from burdock (Arctium lappa Linne). Mutat Res. 1984;129:25–31.
Kada T, Shimoi K. Desmutagens and bio-antimutagens – their modes of action. Bioessays. 1987;7:113–6.
Jain AK, Shimoi K, Nakamura Y, Kada T, Hara Y, Tomita I. Crude tea extracts decrease the mutagenic activity of N-methyl-N’-nitro-N-nitrosoguanidine in vitro and in intragastric tract of rats. Mutat Res. 1989;210:1–8.
Wattenberg LW. Chemoprevention of cancer. Cancer Res. 1985;45:1–8.
Arimoto S, Negishi T, Hayatsu H. Inhibitory effect of hemin on the mutagenic activities of carcinogens. Cancer Lett. 1980;11:29–33.
Negishi T, Arimoto S, Nishizaki C, Hayatsu H. Inhibitory effect of chlorophyll on the genotoxicity of 3-amino-1-methyl-5H-pyrido [4,3-b] indole (Trp-P-2). Carcinogenesis. 1989;10:145–9.
Arimoto S, Hayatsu H. Role of hemin in the inhibition of 3-amino-1-methyl-5H-pyrido [4,3-b] indole (Trp-P-2) and other aminoazaarenes. Mutat Res. 1989;213:217–26.
Ong TM, Whong WZ, Stewart J, Brockman HE. Chlorophyllin: a potent antimutagen against environmental and dietary complex mixtures. Mutat Res. 1986;173:111–5.
Whong WZ, Stewart J, Brockman HE, Ong TM. Comparative antimutagenicity of chlorophyllin and five other agents against aflatoxin B1-induced reversion in Salmonella typhimurium strain TA98. Teratog Carcinog Mutagen. 1988;8:215–24.
Dashwood RH, Breinholt V, Bailey GS. Chemopreventive properties of chlorophyllin: inhibition of aflatoxin B1 (AFB1)-DNA binding in vivo and anti-mutagenic activity against AFB1 and two heterocyclic amines in the Salmonella mutagenicity assay. Carcinogenesis. 1991;12:939–42.
Tachino N, Duo G, Dashwood WM, Yamane S, Larsen R, Dashwood R. Mechanisms of the in vitro antimutagenic action of chlorophyllin against benzo [a]pyrene: studies of enzyme inhibition, molecular complex formation and degradation of the ultimate carcinogen. Mutat Res. 1994;308:191–203.
Jubert C, Bailey GS. Isolation of chlorophylls a and b from spinach by counter-current chromatography. J Chromatogr A. 2007;1140:95–100.
Lynn KS, Cheng ML, Yang HC, Liang YJ, Kang MJ, Chen FL, Shiao MS, Pan WH. Vegetable signatures derived from human urinary metabolomic data in controlled feeding studies. J Proteome Res. 2019;18:159–68. 30517004.
Hayes M, Pottorff M, Kay C, Deynze AV, Osorio-Marin J, Lila MA, Iorrizo M, Ferruzzi MG. In vitro bioaccessibility of carotenoids and chlorophylls in a diverse collection of spinach accessions and commercial cultivars. J Agric Food Chem. 2020;68:3495–505.
Dashwood R, Guo D. Inhibition of 2-amino-3-methylimidazo [4,5-f] quinoline (IQ)-DNA binding by chlorophyllin: studies of enzyme inhibition and molecular complex formation. Carcinogenesis. 1992;13:1121–6.
Dashwood R, Guo D. Antimutagenic potency of chlorophyllin in the Salmonella assay and its correlation with binding constants of mutagen-inhibitor complexes. Environ Mol Mutagen. 1993;22:164–71.
Dashwood R, Yamane S, Larsen R. Study of the forces stabilizing complexes between chlorophylls and heterocyclic amine mutagens. Environ Mol Mutagen. 1996;27:211–8.
Hernaez J, Xu M, Dashwood R. Effects of tea and chlorophyllin on the mutagenicity of N-hydroxy-IQ: studies of enzyme inhibition, molecular complex formation, and degradation/scavenging of the active metabolites. Environ Mol Mutagen. 1997;30:468–74.
Dashwood RH. Protection by chlorophyllin against the covalent binding of 2-amino-3-methylimidazo [4,5-f] quinoline (IQ) to rat liver DNA. Carcinogenesis. 1992;13:113–8.
Guo D, Dashwood R. Inhibition of 2-amino-3-methylimidazo [4,5-f] quinoline (IQ)-DNA binding in rats given chlorophyllin: dose-response and time-course studies in the liver and colon. Carcinogenesis. 1994;15:763–6.
Dashwood RH, Liew C. Chlorophyllin-enhanced excretion of urinary and fecal mutagens in rats given 2-amino-3-methylimidazo [4,5-f]quinoline. Environ Mol Mutagen. 1992;20:199–205.
Guo D, Horio DT, Grove JS, Dashwood RH. Inhibition by chlorophyllin of 2-amino-3-methylimidazo [4,5-f]quinoline-induced tumorigenesis in the male F344 rat. Cancer Lett. 1995;95:161–5.
Hasegawa R, Hirose M, Kato T, Hagiwara A, Boonyaphiphat P, Nagao M, Ito N, Shirai T. Inhibitory effect of chlorophyllin on PhIP-induced mammary carcinogenesis in female F344 rats. Carcinogenesis. 1995;16:2243–6.
Breinholt V, Schimerlik M, Dashwood R, Bailey G. Mechanisms of chlorophyllin anticarcinogenesis against aflatoxin B1: complex formation with the carcinogen. Chem Res Toxicol. 1995;8:506–14.
Breinholt V, Hendricks JD, Pereira C, Arbogast D, Bailey G. Dietary chlorophyllin is a potent inhibitor of aflatoxin B1 hepatocarcinogenesis in rainbow trout. Cancer Res. 1995;55:57–62. 7805041.
Dashwood RH, Negishi T, Hayatsu H, Breinholt V, Hendricks J, Bailey G. Chemopreventive properties of chlorophylls towards AFB1: a review of the antimutagenicity and anticarcinogenicity data in rainbow trout. Mutat Res. 1998;399:245–53.
Breinholt V, Arbogast D, Loveland P, Pereira C, Hendrick J, Bailey G. Chlorophyllin chemoprevention in trout initiated by aflatoxin B (1) bath treatment: an evaluation of reduced bioavailability vs. target organ protective mechanisms. Toxicol Appl Pharmacol. 1999;158:141–51.
Simonich MT, Egner PA, Roebuck BD, Orner GA, Jubert C, Pereira C, Groopman JD, Kensler TW, Dashwood RH, Williams DE, Bailey GS. Natural chlorophyll inhibits aflatoxin B1-induced multi-organ carcinogenesis in the rat. Carcinogenesis. 2007;28:1294–302.
Kensler TW, Groopman JD. Is it time to advance the chemoprevention of environmental carcinogenesis with microdosing trials? Cancer Prev Res. 2009;2:1003–7.
Jubert C, Mata J, Bench G, Dashwood R, Pereira C, Tracewell W, Turteltaub K, Williams D, Bailey G. Effects of chlorophyll and chlorophyllin on low-dose aflatoxin B (1) pharmacokinetics in human volunteers. Cancer Prev Res. 2009;2:1015–22.
Egner PA, Stansbury KH, Snyder EP, Rogers ME, Hintz PA, Kensler TW. Identification and characterization of chlorine (4) ethyl ester in sera of individuals participating in the chlorophyllin chemoprevention trial. Chem Res Toxicol. 2000;13:900–6.
Egner PA, Wang JB, Zhu YR, Zhang BC, Wu Y, Zhang QN, Kuang SY, Gange SJ, Jacobson LP, Helzlsouer KJ, Bailey GS, Groopman JD, Kensler TW. Chlorophyllin intervention reduces aflatoxin-DNA adducts in individuals at high risk for liver cancer. Proc Natl Acad Sci U S A. 2001;98:14601–6.
Arimoto S, Kan-yama K, Rai H, Hayatsu H. Inhibitory effect of hemin, chlorophyllin and related pyrrole pigments on the mutagenicity of benzo [a] pyrene and its metabolites. Mutat Res. 1995;345:127–35.
Park KK, Surh YJ. Chemopreventive activity of chlorophyllin against mouse skin carcinogenesis by benzo [a] pyrene and benzo [a]pyrene-7,8-dihydrodiol-9,10-epoxide. Cancer Lett. 1996;102:143–9.
Katz AK, Carrell HL, Glusker JP. Dibenzo [a,l] pyrene (dibenzo [def,p]chrysene): fjord-region distortions. Carcinogenesis. 1998;19:1641–1648.
Harttig U, Bailey GS. Chemoprotection by natural chlorophylls in vivo: inhibition of dibenzo [a,l]pyrene-DNA adducts in rainbow trout liver. Carcinogenesis. 1998;19:1323–6.
Simonich MT, McQuistan T, Jubert C, Pereira C, Hendricks JD, Schimerlik M, Zhu B, Dashwood RH, Williams DE, Bailey GS. Low-dose dietary chlorophyll inhibits multi-organ carcinogenesis in the rainbow trout. Food Chem Toxicol. 2008;46:1014–24.
McQuistan TJ, Simonich MT, Pratt MM, Pereira CB, Hendricks JD, Dashwood RH, Williams DE, Bailey GS. Cancer chemoprevention by dietary chlorophylls: a 12,000-animal dose-dose matrix biomarker and tumor study. Food Chem Toxicol. 2012;50:342–52.
Castro DJ, Löhr CV, Fischer KA, Waters KM, Webb-Robertson BM, Dashwood RH, Bailey GS, Williams DE. Identifying efficacious approaches to chemoprevention with chlorophyllin, purified chlorophylls and freeze-dried spinach in a mouse model of transplacental carcinogenesis. Carcinogenesis. 2009;30:315–20.
Wu ZL, Chen JK, Ong T, Brockman HE, Whong WZ. Antitransforming activity of chlorophyllin against selected carcinogens and complex mixtures. Teratog Carcinog Mutagen. 1994;14:75–81.
Robins EW, Nelson RL. Inhibition of 1,2-dimethylhydrazine-induced nuclear damage in rat colonic epithelium by chlorophyllin. Anticancer Res. 1989;9:981–5. 2817825.
Nelson RL. Chlorophyllin, as an antimutagen, acts as a tumor promoter in the rat-dimethylhydrazine colon carcinogenesis model. Anticancer Res. 1992;12:737–9. 1622132.
Hirose M, Takahashi S, Ogawa K, Futakuchi M, Shirai T. Phenolics: blocking agents for heterocyclic amine-induced carcinogenesis. Food Chem Toxicol. 1999;37:985–92.
Guo D, Schut HA, Davis CD, Snyderwine EG, Bailey GS, Dashwood RH. Protection by chlorophyllin and indole-3-carbinol against 2-amino-1-methyl-6-phenylimidazo [4,5-b] pyridine (PhIP)-induced DNA adducts and colonic aberrant crypts in the F344 rat. Carcinogenesis. 1995;16:2931–7.
Xu M, Orner GA, Bailey GS, Stoner GD, Horio DT, Dashwood RH. Post-initiation effects of chlorophyllin and indole-3-carbinol in rats given 1,2-dimethylhydrazine or 2-amino-3-methylimidazo [4,5-f]quinoline. Carcinogenesis. 2001;22:309–14.
Blum CA, Xu M, Orner GA, Fong AT, Bailey GS, Stoner GD, Horio DT, Dashwood RH. beta-Catenin mutation in rat colon tumors initiated by 1,2-dimethylhydrazine and 2-amino-3-methylimidazo [4,5-f] quinoline, and the effects of post-initiation treatment with chlorophyllin and indole-3-carbinol. Carcinogenesis. 2001;22:315–20.
Dashwood RH, Xu M, Orner GA, Horio DT. Colonic cell proliferation, apoptosis and aberrant crypt foci development in rats given 2-amino-3-methylimidazo [4,5-f]quinoline. Eur J Cancer Prev. 2001;10:139–45.
Blum CA, Xu M, Orner GA, Diaz GD, Li Q, Dashwood WM, Dashwood RH. Promotion versus suppression of rat colon carcinogenesis by chlorophyllin and chlorophyll: modulation of apoptosis, cell proliferation, and beta-catenin/Tcf signaling. Mutat Res. 2003;523–524:217–23.
Blum CA, Tanaka T, Zhong X, Li Q, Dashwood WM, Pereira C, Xu M, Dashwood RH. Mutational analysis of Ctnnb1 and Apc in tumors from rats given 1,2-dimethylhydrazine or 2-amino-3-methylimidazo [4,5-f]quinoline. Mol Carcinog. 2003;36:195–203.
Wang R, Dashwood WM, Bailey GS, Williams DE, Dashwood RH. Tumors from rats given 1,2-dimethylhydrazine plus chlorophyllin or indole-3-carbinol contain transcriptional changes in beta-catenin that are independent of beta-catenin mutation status. Mutat Res. 2006;601:11–8.
Kawabe M, Futakuchi M, Tamano S, Shirai T, Hirose M. Modifying effects of chitin, chitosan and their related compounds on 2-amino-3,8-dimethylamidazole [4,5-f] quinoxaline (MeIQx) in a rat medium-term hepatocarcinogenesis model, and their post-initiation effects in a female rat 2-stage multi-organ carcinogenesis model. Food Chem Toxicol. 2008;46:2758–63.
Chernomorsky S, Rancourt R, Virdi K, Segelman A, Poretz RD. Antimutagenicity, cytotoxicity and composition of chlorophyllin copper complex. Cancer Lett. 1997;120:141–7.
Dashwood RH. The importance of using pure chemicals in (anti) mutagenicity studies: chlorophyllin as a case in point. Mutat Res. 1997;381:283–6.
Nagai H, Nishiyori T, Daikoku M, Koda A. Immunopharmacological studies of sodium copper chlorophyllin (SCC). Jpn J Pharmacol. 1983:819–28.
Dashwood R, Guo D. Protective properties of chlorophylls against the covalent binding of heterocyclic amines to DNA in vitro and in vivo. Princess Takamatsu Symp. 1995;23:181–9. 8844809.
Dashwood RH. Chlorophylls as anticarcinogens (review). Int J Oncol. 1997;10:721–7 PMID: 21533436.
Dashwood RH. Cancer chemoprevention from the food-borne carcinogen 2-amino-3-methyl-6-phenylimidazo [4,5-b]pyridine. Mutat Res. 1998;405:109–10.
Xu M, Dashwood RH. Chemoprevention studies of heterocyclic amine-induced colon carcinogenesis. Cancer Lett. 1999;143:179–83.
Carter O, Bailey GS, Dashwood RH. The dietary phytochemical alters E-cadherin and beta-catenin expression in human colon cancer cells. J Nutr. 2004;134:3441S–4S.
Cope R, Loehr C, Dashwood R, Kerkvliet NI. Ultraviolet radiation-induced non-melanoma skin cancer in the Crl:SKH1:hr-BR hairless mouse: augmentation of tumor multiplicity by chlorophyllin and protection by indole-3-carbinol. Photochem Photobiol Sci. 2006;5:499–507.
Kwon D, Yoon S, Carter O, Bailey GS, Dashwood RH. Antioxidant and antigenotoxic activities of Angelica keiskei, Oenanthe javanica and Brassica oleracea in the Salmonella mutagenicity assay and in HCT116 human colon cancer cells. Biofactors. 2006;26:231–44.
Subramoniam A, Asha VV, Nair SA, Sasidharan SP, Sureshkumar PK, Rajendran KN, Karunagaran D, Ramalingam K. Chlorophyll revisited: anti-inflammatory activities of chlorophyll a and the inhibition of expression of TNF-α gene by the same. Inflammation. 2012;35:959–66.
Zhang J, Wang W, Yang F, Zhou X, Jin H, Yang PY. Comparative proteomic analysis of drug sodium iron chlorophyllin addition to Hep3B cell line. Analyst. 2012;137:4287–94.
Cheung KK, Chan JY, Fung KP. Antiproliferative effect of pheophorbide a-mediated photodynamic therapy and its synergistic effect with doxorubicin on multiple drug-resistant uterine sarcoma cell MES-SA/Dx5. Drug Chem Toxicol. 2013;36:474–83.
Suryavanshi S, Sharma D, Checker R, Thoh M, Gota V, Sandur SK, Sainis KB. Amelioration of radiation-induced hematopoietic syndrome by an antioxidant chlorophyllin through increased stem cell activity and modulation of hematopoiesis. Free Radic Biol Med. 2015;85:56–70.
Nagini S, Palitti F, Natarajan AT. Chemopreventive potential of chlorophyllin: a review of the mechanisms of action and molecular targets. Nutr Cancer. 2015;67:203–11.
Pietra ED, Simonella F, Bonavida B, Xodo LE, Rapozzi V. Repeated sub-optimal photodynamic treatments with pheophorbide a induce an epithelial mesenchymal transition in prostate cancer cells via nitric oxide. Nitric Oxide. 2015;45:43–53.
Das P, Mandal S, Gangopadhyay S, Das K, Mitra AG, Dasgupta S, Mukhopadhyay A. Antioxidative and anticarcinogenic activities of methylpheophorbide a, isolated from wheat grass (Triticum aestivum Linn.). Nat Prod Res. 2016;30:474–7.
Das J, Samadder A, Mondal J, Abraham SK, Khuda-Bukhsh AR. Nano-encapsulated chlorophyllin significantly delays profession of lung cancer both in in vitro and in vivo models through the activation of mitochondrial signaling cascades and drug-DNA interaction. Environ Toxicol Pharmacol. 2016;46:147–57.
Sarbadhikary P, Dube A. Iodinated chlorin p6 copper complex induces anti-proliferative effect in oral cancer cells through elevation of intracellular reactive oxygen species. Chem Biol Interact. 2017;277:137–44.
Solymosi K, Mysliwa-Kurdziel B. Chlorophylls and their derivatives used in food industry and medicine. Mini Rev Med Chem. 2017;17:1194–222.
Zhuo Z, Song Z, Ma Z, Zhang Y, Xu G, Chen G. Chlorophyllin e6-mediated photodynamic therapy inhibits proliferation and induces apoptosis in human bladder cancer cells. Oncol Rep. 2019;41:2181–93.
Diaz GD, Li Q, Dashwood RH. Caspase-8 and apoptosis-inducing factor mediate a cytochrome c-independent pathway of apoptosis in human colon cancer cells induced by the dietary phytochemical chlorophyllin. Cancer Res. 2003;63:1254–61. 12649185.
Chimploy K, Diaz GD, Li Q, Carter O, Dashwood WM, Mathews CK, Williams DE, Bailey GS, Dashwood RH. E2F4 and ribonucleotide reductase mediate S-phase arrest in colon cancer cells treated with chlorophyllin. Int J Cancer. 2009;125:2086–94.
Krakoff IH, Brown NC, Reichard P. Inhibition of ribonucleotide diphosphate reductase by hydroxyurea. Cancer Res. 1968;28:1559–65 PMID: 4876978.
Liu X, Zhou B, Xue L, Shih J, Tye K, Lin W, Qi C, Chu P, Un F, Wen W, Yen Y. Metastasis-suppressing potential of riboncleotide reductase small subunit p53R2 in human cancer cells. Clin Cancer Res. 2006;12:6337–44.
Aye Y, Li M, Long MJ, Weiss RS. Ribonucleotide reductase and cancer: biological mechanisms and targeted therapies. Oncogene. 2015;34:2011–21.
Mannargudi MB, Deb S. Clinical pharmacology and clinical trials of ribonucleotide reductase inhibitors: is it a viable cancer therapy? J Cancer Res Clin Oncol. 2017;143:1499–529.
Chen G, Luo Y, Warncke K, Sun Y, Yu DS, Fu H, Behera M, Ramalingam SS, Doetsch PW, Duong DM, Lammers M, Curran WJ, Deng X. Acetylation regulates ribonucleotide reductase activity and cancer cell growth. Nat Commun. 2019;10:3212.
Nagini S, Priyadarsini RV, Veeravarmal V, Mishra R. Chlorophyllin abrogates canonical Wnt/beta-catenin signaling and angiogenesis to inhibit the development of DMBA-induced hamster cheek pouch carcinomas. Cell Oncol. 2012;35:385–95.
Parasramka MA, Dashwood WM, Wang R, Abdelli A, Bailey GS, Williams DE, Ho E, Dashwood RH. MicroRNA profiling of carcinogen-induced rat colon tumors and the influence of dietary spinach. Mol Nutr Food Res. 2012;56:1259–69.
Liu Z, Xia S, Wang X, Lan Q, Li P, Xu W, Wang Q, Lu L, Jiang S. Sodium copper chlorophyllin is highly effective against enterovirus (EV) A71 infection by blocking its entry into host cells. ACS Infect Dis. 2020;6:882–90.
Hainer RM. Studies of copper chlorophyllin-odorant systems. Science. 1954;119:609–10.
Young RW, Beregi JS Jr. Use of chlorophyllin in the care of geriatric patients. J Am Geriatr Soc. 1980;28:46–7.
Yamazaki H, Shimizu M. Genetic polymorphism of the flavin-containing monooxygenase 3 (FMO3) associated with trimethylaminuria (fish odor syndrome): observations from Japanese patients. Curr Drug Metab. 2007;8:487–91.
Yamada M, Honma M. Summarized data on genotoxicity tests for designated food additives in Japan. Genes Environ. 2018;40:27 PMID: 30619512.
Wakabayashi K, Nagao M, Esumi H, Sugimura T. Food-derived mutagens and carcinogens. Cancer Res. 1992;52(7 Suppl):2092s–2098s.
Dashwood RH, Suzui M, Nakagama H, Sugimura T, Nagao M. High frequency of beta-catenin (Ctnnb1) mutations in the colon tumors induced by two heterocyclic amines in the F344 rat. Cancer Res. 1998;58:1127–9 PMID: 9515794.
Dashwood RH. Modulation of heterocyclic amine-induced mutagenicity and carcinogenicity: an ‘A-to-Z’ guide to chemopreventive agents, promoters, and transgenic models. Mutat Res. 2002;511:89–112.
Lee HJ, Wu K, Cox DG, Hunter D, Hankinson SE, Willett WC, Sinha R, Cho E. Polymorphisms in xenobiotic metabolizing genes, intakes of heterocyclic amines and red meat, and postmenopausal breast cancer. Nutr Cancer. 2013;65:1122–31.
Sugimura T. In the beginning when the Japanese environmental mutagen society was established. Genes Environ. 2016;38:23.
Nagao M, Tsugane S. Cancer in Japan: prevalence, prevention and the role of heterocyclic amines in human carcinogenesis. Genes Environ. 2016;38:16.
Aoki Y. Evaluation of in vivo mutagenesis for assessing the health risk of air pollutants. Genes Environ. 2017;39:16.
Dashwood RH. Memories of a friend and colleague – Takashi Sugimura. Mutat Res. 2020;786:108337.
Yamada M. In memory of Dr. Takashi Sugimura, 1926–2020. Genes Environ. 2020;42:31.
Chen X, Jia W, Zhu L, Mao L, Zhang Y. Recent advances in heterocyclic aromatic amines: an update on food safety and hazardous control from food processing to dietary intake. Compr Rev Food Sci Food Saf. 2020;19:124–48.
Acknowledgments
The author, first-and-foremost, acknowledges with sincere gratitude the close personal friendship of Professor George S. Bailey from Oregon State University, and the numerous colleagues and trainees from the respective laboratories (and beyond) who contributed to the work discussed in this review article. Apologies are offered for any key reports that might have been omitted from the literature due to space limitations.
Funding
Research from the author’s laboratory that appeared in the review article was supported, in part, by National Institutes of Health (NIH) grants CA090890, CA122959, CA65525 and CA80176.
Author information
Authors and Affiliations
Contributions
The review article was conceived, written and illustrated in its entirety by the author. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All preclinical work reviewed here was approved by the Institutional Animal Care and Use Committee (IACUC), and human translational studies were approved by the Institutional Review Board (IRB).
Consent for publication
Author approves the manuscript submission and it publication.
Competing interests
The author declares that he has no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Dashwood, R.H. Cancer interception by interceptor molecules: mechanistic, preclinical and human translational studies with chlorophylls. Genes and Environ 43, 8 (2021). https://doi.org/10.1186/s41021-021-00180-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41021-021-00180-8