
Abstract
Background
Glioma is a common malignant brain tumor. The purpose of this study was to investigate the role of the transcription factor SPI1 in glioma.
Methods
SPI1 expression in glioma was identified using qRT-PCR and Western blotting. Cell proliferation was assessed using the CCK8 assay. Transwell and wound healing assays were utilized to evaluate cell migration. Additionally, cell cycle and apoptosis were detected using flow cytometry.
Results
We observed that the expression level of SPI1 was up-regulated in glioma tissues, compared to normal tissues. Furthermore, we found that SPI1 is able to promote proliferation and migration of glioma cells in vitro. Flow cytometry results demonstrate that, compared to si-NC cells, si-SPI1 cells stagnated in the G1 phase, and down-regulation of SPI1 expression is able to increase rates of apoptosis. Double luciferase activity and chromatin immunoprecipitation assay results indicated that SPI1 can bind to the promoter sites and promote the proliferation and migration of glioma cells by regulating the expression of oncogenic PAICS.
Conclusions
Our results suggest that SPI1 can promote proliferation and migration of glioma. Furthermore, SPI1 can be utilized as a potential diagnostic marker and therapeutic target for glioma.
Similar content being viewed by others


Background
Glioma is a disease that seriously endangers human health. Thus far, the pathogenesis of glioma remains unclear. Clinically, patients suffer from the pain caused by radiotherapy and chemotherapy, which causes serious economic and mental burden to society and family.
The transcription factor SPI1 is an important regulatory protein that plays an extremely important role in blood cell differentiation, immune cell differentiation, as well as tumor development [1, 2]. To date, no studies have reported on the relationship between SPI1 Phosphoribosylaminoimidazole Carboxylase and Phosphoribosylaminoimidazolesuccinocarboxamide Synthase (PAICS) in glioma. Numerous studies demonstrated that PAICS can promote the proliferation and migration of many tumors [3,4,5,6,7,8]. Our previous study also found that PAICS plays an oncogene role in glioma [9]. In this project, we intend to further study the transcriptional regulation of SPI1 and PAICS through luciferase assay, gene transfection technology, Western blot and other techniques. Our goal was to explore the role of SPI1 in the occurrence and development of glioma with regards to regulation of PAICS, and provide a new direction for further exploration of glioma pathogenesis.
Methods
Clinical specimens and cell lines
Human gliomas and surrounding normal brain tissues requiring decompression were collected from the Xinxiang Central Hospital in Henan Province. Written informed consent was obtained from all patients or their relatives. All work was granted approval by the Medical Ethics Committee of Xinxiang Central Hospital. Tissue specimens were frozen in liquid nitrogen and stored at -80°C. The glioma cell line used in this study was obtained from ATCC, and cultured in RPMI 1640 (Hyclone) containing 10% Fetal Bovine Serum (FBS) (Gibco) at 37°C and 5% CO2.
RNA extraction and qRT-PCR
RNA was extracted using the TRIZol reagent (Takara). The cDNA was synthesized using the Takara reverse transcription kit. Real-time quantitative RT-PCR experiments were performed using cDNA as the template. SYBR Green I (Takara) was added, and the primers were mixed. The mixture was run on Applied Biosystems 7500/7500 Fast (Applied Biosystems), with GAPDH as the internal control. All experiments were repeated three times. SPI1 reaction conditions were as follows: 95°C for 5 min, 40 cycles at 95°C for 1 min, Tm of 30 s, and then 72°C for 34 s. The ΔCt method was used to calculate relative expression of SPI1. The ΔCt values were used to compare the expression level of SPI1 in tumor vs the control group. ΔCt = CtSPI1 – CtGAPDH. The SPI1 primers are listed as follows. The forward primer was 5′- GCGACCATTACTGGGACTTCC - 3′ and the reverse primer was 5′- GGGTATCGAGGACGTGCAT -3′. GAPDH was used as an internal control. The primers for GAPDH were 5′- GACTCATGACCACAGTCCATGC - 3′ and 5′- AGAGGCAGGGATGATGTTCTG -3′.
Construction of a SPI1-knockdown cell lines and transfection
The SPI1 siRNA was purchased from Gene Pharmaceutical (Shanghai, China). The nucleotide sequence for the SPI1 siRNA is 5'- GAGAGCTTCGCCGAGAACAACTTCA-3'. Transfection of siRNA oligonucleotides and plasmids was conducted with Lipofectamine 2000, which was purchased from Invitrogen, ThermoFisher Scientific (Catalog No.11668019, Massachusetts, USA).
Transwell assays
Cultured cells in the logarithmic phase were digested by trypsin in order to make single cell suspension waves. The cell concentration was adjusted to (1-2)×l05/mL. Next, 100 μl of the cell suspension was added to the upper chamber, and then 500 μl containing 10% of newborn bovine serum was added to the lower chamber. The cells were cultured for 48h at 37°C. The small chamber was taken out, fixed with 4% paraformaldehyde for 20 min, and stained with Giemsa for 15 min. Under the microscope, 5 fields were randomly chosen to count the number of cells that passed through the membrane in a 200X microscope. The influence of changes in SPI1 expression on cell migration was compared. The transwell chamber was purchased from Corning, Inc., USA.
Wound healing assay
Cultured cells in the logarithmic state were seeded onto a 6-well plate with 8x105 cells per well at 1-2 days before the experiment. The cells were incubated at 37°C in a 5% CO2 incubator. When the cells were grown to approximately 90% confluency, a 200 μl sterile spear was used to create a scratch on the 6-well plate. The liquid was then gently shaken and discarded to remove the floating cells. Pictures were taken under an inverted microscope at 10-times the field of view. Cell migration at the scratch was recorded at different time points.
Cell cycle detection
The transfected cells were collected and centrifuged at 4°C. The supernatant was discarded, and fixed with pre-cooled 70% ethanol for 24 h. Then, the cells were washed with PBS. Next, 10 μl of RNase A solution was added to each sample, as well as 40 μl of PBS, according to the instructions of the cell cycle detection kit. The mixture was placed in the water bath at 37°C for 30 min. The red fluorescence at the excitation wavelength of 488nm was recorded by flow cytometry.
Cell apoptosis detection
Cell apoptosis was evaluated by Annexin V/PI double labeling. The cells were digested and collected, washed twice with PBS, and made resuspended into a single cell suspension. Cell suspension was then incubated with Annexin V-FITC and PI at room temperature, and placed in the dark for 15 min. Flow cytometry was utilized to determine the results, and the CellQuest analysis software helped analyze the results.
Double luciferase activity assay
The Dual-Luciferase ReporterAssay System (Catalog No. E1910) of Promega was utilized to quantify dual luciferase activity. Additionally, 48h after the plasmid was transfected, the medium was removed and washed twice with PBS. Next, 65 μl of 1× Passive Lysis Buffer (PLB) was added to each well. Then, the cell lysates were collected after gentle vibration at room temperature for 15 min. We utilized an automatic luminous detector, which required adding 20 μl of the cell lysate to each hole, and then adding 100 μl of LAR II to automatically detect the fluorescence value of firefly (F). After adding a 100 μl of the STOP&Glo@Reagent, the fluorescence value (R) was determined immediately. After reading the value, the data was saved for analysis of the detection results.
Chromatin immunoprecipitation (ChIP)
ChIP assays were carried out according to the EZ-CHIP kit (Millipore, Temecula, CA, USA). The anti-SPI1 (Abclone) antibodies were utilized to precipitate the DNA-protein complex. The immunoprecipitated DNA was evaluated by PCR. Primers that were specific to the PAICS promoter containing E-box were 5’-TAAAGTTCATGGAAGCGAGGTG-3’ (forward) and 5’- TCACTCTGGGACTCGTGGG-3’ (reverse).
Statistical analysis
Data was analyzed using the SPSS 20.0 statistical software. Quantitative data was presented as mean ± SD of at least three independent experiments. The differences between the independent experimental groups were assessed using a two-tailed Student's t-test. Differences were considered significant if p < 0.05*, p < 0.01**, p < 0.001***.
Results
Increasing SPI1 levels correlate with glioma progression
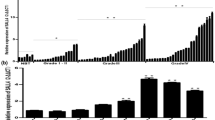
In order to investigate the role of SPI1 in glioma tumorigenesis, we determined the expression of SPI1 in 10 paired glioblastoma (GBM) tissues and adjacent normal counterparts by qRT-PCR. The expression of SPI1 was normalized to GAPDH, and results were presented as fold-change in tumor tissues relative to matched adjacent normal tissues. Formula =2-ΔΔCt was utilized to calculate relative expression of SPI1 in tissues. Paired-samples t-test was used to determine the significance of the ΔCt values between the tumor and control group. The results revealed that SPI1 expression was increased in 9 of 10 GBM specimens (P < 0.05) (Fig. 1a, b). Additionally, western blot assay helped determine the protein expression of SPI1 in three paired GBM tissues. The results demonstrated that protein expression of SPI1 in GBM tissues was higher compared to paired adjacent normal tissues (P<0.05) (Fig. 1c, d). In the online GEPIA database (http://gepia.cancer-pku.cn/) and UACLAN database ((http://ualcan.path.uab.edu/), we analyzed the expression and survival of SPI1 [10, 11]. The results indicated that expression of SPI1 was significantly up-regulated in the tumor, and was associated with a lower survival rate in GBM (Fig. 1e-g). The protein expression of SPI1 in glioma tissues was validated through the use of data from Proteinatlas Version 19.3 (http://www.proteinatlas.org) (Supplementary Fig. S1) [12,13,14].

Expression levels of SPI1 in glioblastoma tissues. a RT-qPCR analysis of SPI1 expression in glioblastoma tissues compared with the control brain tissues. Error bars indicate the mean ± standard deviation of three independent experiments. (N: normal brain tissue, T: glioma tissue, green line represents T/N=1). b The value of ΔCt was used to show the expression level of SPI1 (ΔCt =Ct (SPI1) − Ct (GAPDH)) in the 10 paired human glioblastoma tissues and control brain tissues (P < 0.01). c Western blot analysis of SPI1 protein expression in three pair glioblastoma tissues compared with the normal tissues. The differences between tumor group and normal group were tested by using independent-samplest-test. d Protein expression level of SPI1 was normalized to GAPDH and quantified using Image J. Error bars represent the mean ± SD of 3 different tests. ***, p < 0.001. e Expression levels of SPI1 mRNA with glioma tissues and control tissues in GEPIA database . *, p < 0.05. f Expression levels of SPI1 protein with glioma tissues and control tissues in UALCAN database. g Survival curve of low SPI1 expression group and high SPI1 expression group
SPI1 promotes glioma cell lines proliferation and migration in vitro
In order to understand SPI1 functionality in glioma progression, we knocked down PAICS expression with siRNAs in the U87 and U251 cells, respectively. The qRT-PCR and western blot assays were performed to determine the expression of SPI1. The results demonstrated that the mRNA and protein expression of SPI1 was reduced (Fig. 2a, b). Furthermore, CCK8 results showed that the knockdown of SPI1 reduced cell growth (Fig. 2c). In addition, transwell and wound healing assay were used to detect motility, the results of which revealed that repression of SPI1 attenuates migration of U87 and U251 cells (Fig. 2d, e).

Inhibition of SPI1 in glioma cells weakened the cell growth and metastasis in vitro. a RT-qPCR analysis of SPI1 mRNA levels after the transfection of the si-NC or si-SPI1 small interfering RNAs. SPI1 expression was normalized to GAPDH, Error bars indicate mean ± SD of 3 independent experiments.**, p< 0.01; ***, p< 0.001. b Western blot analysis of SPI1 protein levels after the transfection of the si-NC or si-SPI1 small interfering RNAs. SPI1 expression was normalized to β-Actin, Error bars indicate mean ± SD of 3 independent experiments. ***, p< 0.001. c Knockdown of SPI1 inhibited cell proliferation on the basis of CCK8 assays. Error bars represent the mean ± SD of 5 independent experiments. **, p< 0.01;***, p< 0.001. d Inhibition of SPI1 decreased cell migration as determined by transwell assays. The bar chart represents the migration cell numbers. Error bars represent the mean ± SD of 5 different field. ***, p< 0.001. e Inhibition of SPI1 decreased cell migration as determined by wound healing assay. The bar chart represents the percentage of distance at 24h or 48h divided by the distance at 0h. Date are presented as mean ± SD of 3 independent experiments. ***, p< 0.001
Inhibition of SPI1 suppresses cell cycle and apoptosis in glioma cell lines
Flow cytometry revealed that knock down in the expression of SPI1 in U87 and U251 cells increased the proportion of cells in G0/G1 phase, and they decreased the proportion of glioma cells in the S phase (Fig. 3a). Furthermore, we discovered that a decrease in the expression of SPI1 increased the apoptotic rate of glioma cells (Fig. 3b).

Knockdown of SPI1 in glioma cells inhibit cell cycle and promote apoptosis in vitro. a Cell cycle was analyzed by flow cytometry after knocking down PAICS. The results showed that PAICS depletion led G1 arrest. b Cell apoptosis was analyzed by flow cytometry after knocking down PAICS. The results showed that PAICS depletion leading to an increased rate of apoptosis
SPI1 is a transcriptional regulator of PAICS
We analyzed the potential transcription factor binding sites in the promoter regions of PAICS from the online database JASPAR (http://jaspar.genereg.net/), hTFTARGET (https://bio.tools/hTFtarget) and HumanTFDB (http://bioinfo.life.hust.edu.cn/HumanTFDB#!/). We found that the promoter regions could be possible binding sites recognized by SPI1 (Supplementary Fig. S2) [15,16,17]. We observed that SPI1 effectively stimulated the luciferase activity of PAICS promoter in U87 and U251 cells (Fig. 4a). Moreover, ChIP results indicated that the transcription factor SPI1 can bind to the PAICS promoter region (Fig. 4b). Finally, we conducted knockdown of SPI1, which led to a decrease in the expression of PAICS in U87 and U251 cells (Fig. 4c). These results indicate that SPI1 can directly bind to the promoter region and regulate the expression of PAICS.

SPI1 binds to the promoter regions of PAICS and regulates PAICS expression. a Schematic of the PAICS promoter luciferase construct is depicted with the locations of the GGGAAG element and the sequences of mutation. Relative luciferase activities of the Wt or Mut GGGAAG regions in U87 and U251 cells were examined after transfecting with SPI1 48 hour later. The values of PGL3-WT and PGL3-Mut were relative to PGL3-basic. Data are mean ± SD for triplicate samples. ***, p < 0.001. b SPI1 binding at the promoter region of PAICS containing the GGGAAG element was assessed by CHIP assay. c PAICS RNA levels after transfecting the SPI1 interference fragment into the U87 and U251 cells. PAICS expression was normalized to GAPDH. Error bars indicate mean ±SD of 3 independent experiments. *, p < 0.05; ** p < 0.01; ***, p < 0.001
SPI1 promotes glioma cell proliferation and migration through PAICS
Next, we sought to evaluate whether SPI1 can regulate cell proliferation and migration through PAICS. We used Western blot to detect the protein expression of SPI1 and PAICS. Meanwhile, we conducted the CCK8 cell experiment and transwell assay to detect cell proliferation and migration. The results demonstrated that PAICS was able to reverse the decrease in proliferation and migration of glioma cells induced by SPI1 interference (Fig. 5a, b).

SPI1 promotes glioma cell proliferation and migration through PAICS. a PAICS can reverse the reduction of cell proliferation induced by SPI1 interference on the basis of CCK8 assays. Error bars represent the mean ± SD of 3 independent experiments. **, p< 0.01;***, p< 0.001. b PAICS can reverse the reduction of cell migration induced by SPI1 interference on the basis of transwell assays. The bar chart represents the migration cell numbers. Error bars represent the mean ± SD of 3 different field. ***, p< 0.001
Discussion
Glioma is one of the most common brain tumors that is associated with high mortality. The SPI1 transcription factor belongs to ETS (e-twenty six) transcription factor family [18, 19], which is one of the largest families of transcription factors, comprised of 28 genes [20]. All ETS family members have a highly conserved DNA binding domain, known as the ETS domain, which has a helix-to-helix flank that binds to DNA binding sites with the GGA(A/T) sequence [21, 22]. The ETS family has many functions that are related to the development of different tissues and cancer, including regulation of cell differentiation, cell cycle regulation, cell migration, cell proliferation, apoptosis, angiogenesis, and other functions [23, 24]. Many ETSs have been associated with cancer, such as ERG (ETS-Related Gene), through gene fusion. When ERG transcription factors fuse with the EWS Gene, it causes Ewing's sarcoma [25, 26]. As a member of the ETS family, SPI1 plays an important role in the development of bone marrow and B lymphocytes, as well as activation of corresponding gene expression in tumors [27]. SPI1 binds to a PU-box sequence near the promoter of target genes, a purine-rich DNA sequence, and coregulates its expression in concert with other transcription factors and cofactors. The SPI1 protein can also bind RNA in order to regulate selective splicing of target genes [28].
In breast cancer, lung cancer and cervical cancer, SPI1 plays a role in promoting cancer [29]. In glioma, results have shown that SPI1 plays a cancer-promoting role [30, 31], but the functional verification and mechanism of SPI1 have not yet been reported. In this study, we investigated expression, function and mechanism of SPI1 in glioma. Compared to the normal group, the SPI1 mRNA and protein are up-regulated in glioma tissues. Moreover, we discovered that suppression of SPI1 decreases glioma cell growth and migration. Therefore, the results demonstrated that SPI1 exerts an oncogenic effect to promote cell growth and migration in glioma.
The mechanism of how SPI1 promotes glioma progression remains uncertain. One study has reported that PAICS can promote tumor proliferation and migration in neuroblastoma [32], though it has also been shown to play an important role in a variety of tumors [4,5,6]. Our previous study also found that PAICS plays an oncogene role in glioma [9]. Bioinformatics analysis indicated that SPI1 can bind to the PAICS promoter region. We speculate that SPI1 can play a role in promoting cancer through PAICS. Experimental results indicated that SPI1 can regulate expression of PAICS, and dual luciferase reporter assay verified binding of SPI1 with the PAICS promoter region. Through the rescue experiment, we also validated that SPI1 can promote proliferation and migration of tumor cells in glioma through PAICS.
Conclusion
In conclusion, our results indicate that SPI1 is a tumor promoting gene. When inhibited, it may inhibit proliferation and migration of glioma cells. The role of SPI1 in the development of glioma is related to regulation of PAICS. Our results indicate that SPI1 may be a promising target for the treatment of glioma.
Availability of data and materials
All data relevant to the study are included in the article. The datasets used or analyzed during the current study are available.
Abbreviations
- GBM:
-
Glioblastoma
- ETS:
-
E-twenty six
- BBB:
-
Blood-brain barrier
- ERG:
-
ETS Related Gene
- PAICS:
-
Phosphoribosylaminoimidazole Carboxylase And Phosphoribosylaminoimidazolesuccinocarboxamide Synthase
- PLB:
-
Passive Lyrsis Buffer
- ChIP:
-
Chromatin immunoprecipitation
- FBS:
-
Fetal Bovine Serum
References
Guo WQ, Chen YG, Shi RZ, He K, Wang JF, Shao JH, et al. 20(S)-Protopanaxdiol Suppresses the Abnormal Granule-Monocyte Differentiation of Hematopoietic Stem Cells in 4T1 Breast Cancer-Bearing Mouse. Evid Based Complement Alternat Med. 2020;2020:8747023. https://doi.org/10.1155/2020/8747023.
Gupta P, Gurudutta GU, Saluja D, Tripathi RP. PU.1 and partners: regulation of haematopoietic stem cell fate in normal and malignant haematopoiesis. J Cell Mol Med. 2009;13(11-12):4349–63. https://doi.org/10.1111/j.1582-4934.2009.00757.x.
Meng M, Chen Y, Jia J, Li L, Yang S. Knockdown of PAICS inhibits malignant proliferation of human breast cancer cell lines. Biol Res. 2018;51(1):24. https://doi.org/10.1186/s40659-018-0172-9.
Agarwal S, Chakravarthi B, Kim HG, Gupta N, Hale K, Balasubramanya SAH, et al. PAICS, a De Novo Purine Biosynthetic Enzyme, Is Overexpressed in Pancreatic Cancer and Is Involved in Its Progression. Transl Oncol. 2020;13(7):100776. https://doi.org/10.1016/j.tranon.2020.100776.
Zhou S, Yan Y, Chen X, Wang X, Zeng S, Qian L, et al. Roles of highly expressed PAICS in lung adenocarcinoma. Gene. 2019;692:1–8. https://doi.org/10.1016/j.gene.2018.12.064.
Agarwal S, Chakravarthi B, Behring M, Kim HG, Chandrashekar DS, Gupta N, et al. PAICS, a Purine Nucleotide Metabolic Enzyme, is Involved in Tumor Growth and the Metastasis of Colorectal Cancer. Cancers (Basel). 2020;12(4). https://doi.org/10.3390/cancers12040772.
Chakravarthi B, Rodriguez Pena MDC, Agarwal S, Chandrashekar DS, Hodigere Balasubramanya SA, Jabboure FJ, et al. A Role for De Novo Purine Metabolic Enzyme PAICS in Bladder Cancer Progression. Neoplasia. 2018;20(9):894–904. https://doi.org/10.1016/j.neo.2018.07.006.
Chakravarthi BV, Goswami MT, Pathi SS, Dodson M, Chandrashekar DS, Agarwal S, et al. Expression and Role of PAICS, a De Novo Purine Biosynthetic Gene in Prostate Cancer. Prostate. 2017;77(1):10–21. https://doi.org/10.1002/pros.23243.
Du B, Zhang Z, Di W, Xu W, Yang L, Zhang S, et al. PAICS is related to glioma grade and can promote glioma growth and migration. J Cell Mol Med. 2021;25(16):7720–33. https://doi.org/10.1111/jcmm.16647.
Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):W98–W102. https://doi.org/10.1093/nar/gkx247.
Chandrashekar DS, Bashel B, Balasubramanya SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi BVSK, et al. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia. 2017;19(8):649–58. https://doi.org/10.1016/j.neo.2017.05.002.
Uhlen M, Zhang C, Lee S, Sjostedt E, Fagerberg L, Bidkhori G, et al. A pathology atlas of the human cancer transcriptome. Science. 2017;357(6352). https://doi.org/10.1126/science.aan2507.
Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347(6220):1260419. https://doi.org/10.1126/science.1260419.
Thul PJ, Akesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, et al. A subcellular map of the human proteome. Science. 2017;356(6340). https://doi.org/10.1126/science.aal3321.
Fornes O, Castro-Mondragon JA, Khan A, van der Lee R, Zhang X, Richmond PA, et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020;48(D1):D87–92. https://doi.org/10.1093/nar/gkz1001.
Zhang Q, Liu W, Zhang HM, Xie GY, Miao YR, Xia M, et al. hTFtarget: A Comprehensive Database for Regulations of Human Transcription Factors and Their Targets. Genomics Proteomics Bioinformatics. 2020;18(2):120–8. https://doi.org/10.1016/j.gpb.2019.09.006.
Hu H, Miao YR, Jia LH, Yu QY, Zhang Q, Guo AY. AnimalTFDB 3.0: a comprehensive resource for annotation and prediction of animal transcription factors. Nucleic Acids Res. 2019;47(D1):D33–8. https://doi.org/10.1093/nar/gky822.
Verbiest T, Bouffler S, Nutt SL, Badie C. PU.1 downregulation in murine radiation-induced acute myeloid leukaemia (AML): from molecular mechanism to human AML. Carcinogenesis. 2015;36(4):413–9. https://doi.org/10.1093/carcin/bgv016.
Hsing M, Wang Y, Rennie PS, Cox ME, Cherkasov A. ETS transcription factors as emerging drug targets in cancer. Med Res Rev. 2020;40(1):413–30. https://doi.org/10.1002/med.21575.
Cooper CD, Newman JA, Gileadi O. Recent advances in the structural molecular biology of Ets transcription factors: interactions, interfaces and inhibition. Biochem Soc Trans. 2014;42(1):130–8. https://doi.org/10.1042/BST20130227.
Szymczyna BR, Arrowsmith CH. DNA binding specificity studies of four ETS proteins support an indirect read-out mechanism of protein-DNA recognition. J Biol Chem. 2000;275(37):28363–70. https://doi.org/10.1074/jbc.M004294200.
Adamo P, Ladomery MR. The oncogene ERG: a key factor in prostate cancer. Oncogene. 2016;35(4):403–14. https://doi.org/10.1038/onc.2015.109.
Fry EA, Inoue K. Aberrant expression of ETS1 and ETS2 proteins in cancer. Cancer Rep Rev. 2018;2(3). https://doi.org/10.15761/CRR.1000151.
Fry EA, Mallakin A, Inoue K. Translocations involving ETS family proteins in human cancer. Integr Cancer Sci Ther. 2018;5(4). https://doi.org/10.15761/ICST.1000281.
Kedage V, Selvaraj N, Nicholas TR, Budka JA, Plotnik JP, Jerde TJ, et al. An Interaction with Ewing's Sarcoma Breakpoint Protein EWS Defines a Specific Oncogenic Mechanism of ETS Factors Rearranged in Prostate Cancer. Cell Rep. 2016;17(5):1289–301. https://doi.org/10.1016/j.celrep.2016.10.001.
Grunewald TGP, Cidre-Aranaz F, Surdez D, Tomazou EM, de Alava E, Kovar H, et al. Ewing sarcoma. Nat Rev Dis Primers. 2018;4(1):5. https://doi.org/10.1038/s41572-018-0003-x.
Somasundaram R, Prasad MA, Ungerback J, Sigvardsson M. Transcription factor networks in B-cell differentiation link development to acute lymphoid leukemia. Blood. 2015;126(2):144–52. https://doi.org/10.1182/blood-2014-12-575688.
Dekoninck A, Calomme C, Nizet S, de Launoit Y, Burny A, Ghysdael J, et al. Identification and characterization of a PU.1/Spi-B binding site in the bovine leukemia virus long terminal repeat. Oncogene. 2003;22(19):2882–96. https://doi.org/10.1038/sj.onc.1206392.
Elton TS, Selemon H, Elton SM, Parinandi NL. Regulation of the MIR155 host gene in physiological and pathological processes. Gene. 2013;532(1):1–12. https://doi.org/10.1016/j.gene.2012.12.009.
Lin X, Jiang T, Bai J, Li J, Wang T, Xiao J, et al. Characterization of Transcriptome Transition Associates Long Noncoding RNAs with Glioma Progression. Mol Ther Nucleic Acids. 2018;13:620–32. https://doi.org/10.1016/j.omtn.2018.10.009.
Xu Y, Gu S, Bi Y, Qi X, Yan Y, Lou M. Transcription factor PU.1 is involved in the progression of glioma. Oncol Lett. 2018;15(3):3753–9. https://doi.org/10.3892/ol.2018.7766.
Cheung CHY, Hsu CL, Tsuei CY, Kuo TT, Huang CT, Hsu WM, et al. Combinatorial targeting of MTHFD2 and PAICS in purine synthesis as a novel therapeutic strategy. Cell Death Dis. 2019;10(11):786. https://doi.org/10.1038/s41419-019-2033-z.
Acknowledgements
Not applicable
Funding
This work is supported by National Natural Science Foundation of China (Grant No.81802470), Joint construction project of Henan Medical Science and technology research plan (Grant No. LHGJ20190452), the Doctoral Scientific Research Foundation of Xinxiang Medical University (Grant No.XYBSKYZZ201632), Natural Science Foundation of Henan Province (Grant No. 202300410326).
Author information
Authors and Affiliations
Contributions
BD conceived and designed the experiments, prepared figures and approved the final draft. WG performed the experiments, authored or reviewed drafts of the paper, and approved the final draft. YQ performed the experiments and prepared figures. JZ analyzed the data, reviewed drafts of the paper, and approved the final draft. ZZ conceived and designed the experiments, analyzed the data, prepared figures and approved the final draft. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Informed consent was obtained from patients to collect tissue samples for the study, and the study was reviewed and approved by the Ethics Committee of Xinxiang Central Hospital (No. 2019-086).
Consent for publication
Not applicable
Competing interests
The authors declare that they have no conflict of interest.
Supplementary Information
Additional file 1: Figure S1.
Expression levels of SPI1 in glioblastoma tissues were predited by online database HPA. Use the Human Protein Atlas (HPA) online database to analyze the protein expression of SPI1. Above is the IHC results of normal brain tissues. Below is IHC results of glioma tissues.
Additional file 2: Figure S2.
Prediction of the potential transcript factor binding sites in the promoter regions of PAICS from online database JASPAR, hTFTARGET and HumanTFDB. a Prediction of transcription factor binding site of JASPAR database. b Prediction of transcription factor binding site of hTFTARGET database. c Prediction of transcription factor binding site of HumanTFDB database.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
{kind=link}
Cite this article
Du, B., Gao, W., Qin, Y. et al. Study on the role of transcription factor SPI1 in the development of glioma. Chin Neurosurg Jl 8, 7 (2022). https://doi.org/10.1186/s41016-022-00276-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41016-022-00276-2